
In some ways, it’s surprising that carbon NMR is even possible. After all, 12C, the most abundant carbon isotope, has no nuclear spin and can’t be seen by NMR. Carbon-13 is the only naturally occurring carbon isotope with a nuclear spin, but its natural abundance is only 1.1%. Thus, only about 1 of every 100 carbon atoms in an organic sample can be observed by NMR. The problem of low abundance has been overcome, however, by the use of signal averaging and Fourier-transform NMR (FT–NMR). Signal averaging increases instrument sensitivity, and FT–NMR increases instrument speed.
The low natural abundance of 13C means that any individual NMR spectrum is extremely “noisy.” That is, the signals are so weak that they are cluttered with random background electronic noise, as shown in Figure 13.17a. If, however, hundreds or thousands of individual runs are added together by a computer and then averaged, a greatly improved spectrum results (Figure 13.17b). Background noise, because of its random nature, increases very slowly as the runs are added, while the nonzero signals stand out clearly. Unfortunately, the value of signal averaging is limited when using the method of NMR spectrometer operation described in Section 13.2, because it takes about 5 to 10 minutes to obtain a single spectrum. Thus, a faster way to obtain spectra is needed if signal averaging is to be used.
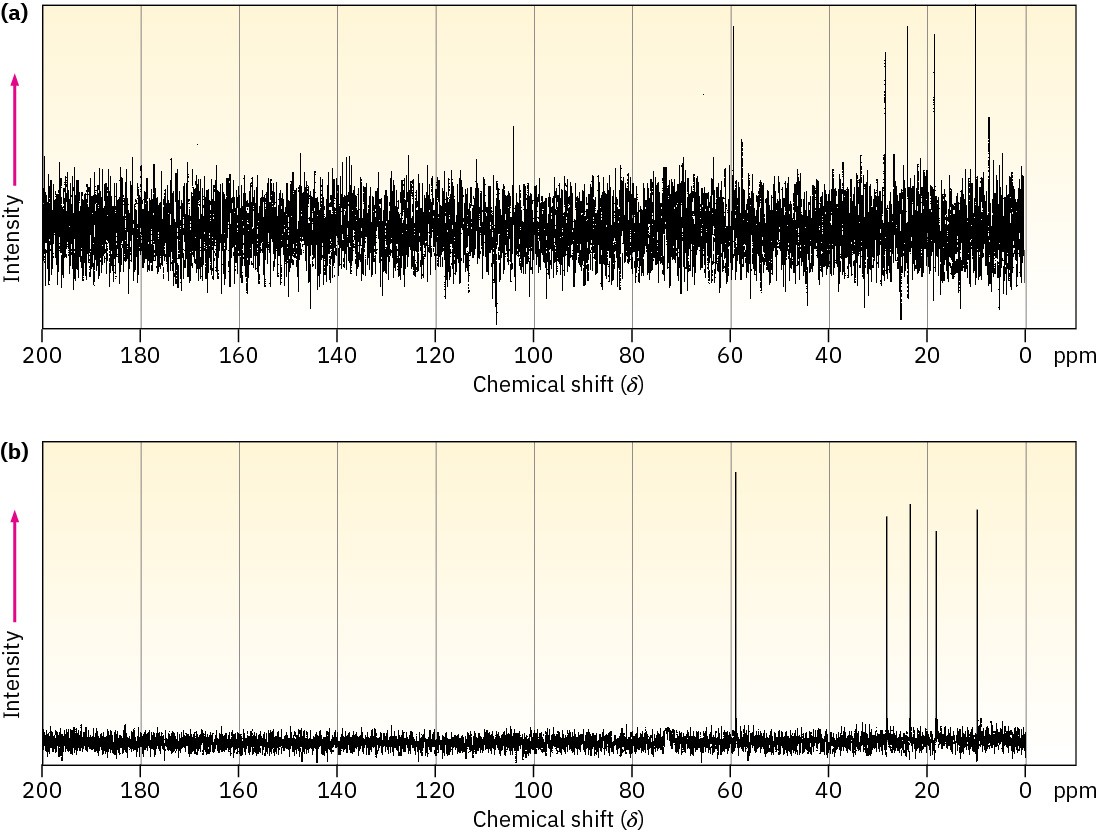
Figure 13.17 Carbon-13 NMR spectra of 1-pentanol. Spectrum (a) is a single run, showing the large amount of background noise. Spectrum (b) is an average of 200 runs.
In the method of NMR spectrometer operation described in Section 13.2, the rf frequency is held constant while the strength of the magnetic field is varied so that all signals in the spectrum are recorded sequentially. In the FT–NMR technique used by modern spectrometers, however, all the signals are recorded simultaneously. A sample is placed in a magnetic field of constant strength and is irradiated with a short pulse of rf energy that covers the entire range of useful frequencies. All 1H or 13C nuclei in the sample resonate at once, giving a complex, composite signal that is mathematically manipulated using so-called Fourier transforms and then displayed in the usual way. Because all resonance signals are collected at once, it takes only a few seconds rather than a few minutes to record an entire spectrum.
Combining the speed of FT–NMR with the sensitivity enhancement of signal averaging is what gives modern NMR spectrometers their power. Literally thousands of spectra can be taken and averaged in a few hours, resulting in sensitivity so high that a 13C NMR spectrum can be obtained from less than 0.1 mg of sample and a 1H spectrum can be recorded from only a few micrograms.
One further question needs to be answered before moving forward with our discussion of 13C NMR. Why is spin–spin splitting seen only for 1H NMR? Why is there no splitting of carbon signals into multiplets in 13C NMR? After all, you might expect that the spin of a given 13C nucleus would couple with the spin of an adjacent magnetic nucleus, either 13C or 1H.
No coupling of a 13C nucleus with nearby carbons is seen because their low natural abundance makes it unlikely that two 13C nuclei will be adjacent. No coupling of a 13C nucleus with nearby hydrogens is seen because 13C spectra are normally recorded using broadband decoupling. At the same time that the sample is irradiated with a pulse of rf energy to cover the carbon resonance frequencies, it is also irradiated by a second band of rf energy covering all the hydrogen resonance frequencies. This second irradiation makes the hydrogens spin-flip so rapidly that their local magnetic fields average to zero and no coupling with carbon spins occurs.
