
All the structural models in this book are computer-drawn. To make sure they accurately represent bond angles, bond lengths, torsional interactions, and steric interactions, the most stable geometry of each molecule has been calculated on a desktop computer using a commercially available molecular mechanics program based on work by Norman Allinger at the University of Georgia.
The idea behind molecular mechanics is to begin with a rough geometry for a molecule and then calculate a total strain energy for that starting geometry, using mathematical equations that assign values to specific kinds of molecular interactions. Bond angles that are too large or too small cause angle strain; bond lengths that are too short or too long cause stretching or compressing strain; unfavorable eclipsing interactions around single bonds cause torsional strain; and nonbonded atoms that approach each other too closely cause steric, or van der Waals, strain.
𝐸total = 𝐸bond stretching + 𝐸angle strain + 𝐸torsional strain + 𝐸van der Waals
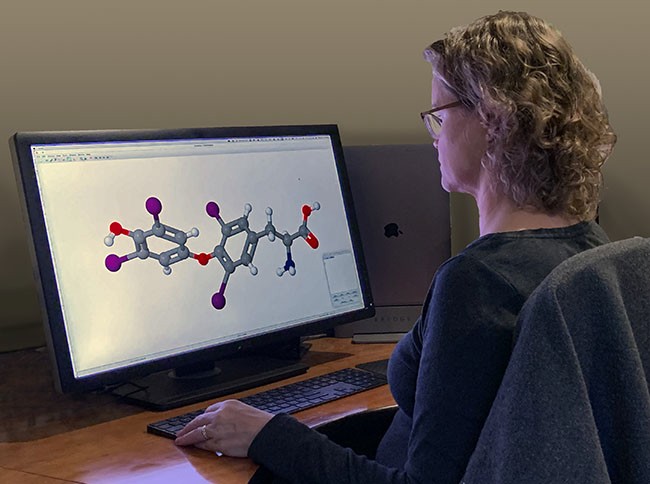
Figure 4.19 Computer programs make it possible to accurately represent molecular geometry. (credit: “Molecular geometry” by Jane Whitney/Flickr, CC BY 4.0)
After calculating a total strain energy for the starting geometry, the program automatically changes the geometry slightly in an attempt to lower strain—perhaps by lengthening a bond that is too short or decreasing an angle that is too large. Strain is recalculated for the new geometry, more changes are made, and more calculations are done. After dozens or hundreds of iterations, the calculation ultimately converges on a minimum energy that corresponds to the most favorable, least strained conformation of the molecule.
Similar calculations have proven to be particularly useful in pharmaceutical research, where a complementary fit between a drug molecule and a receptor molecule in the body is often the key to designing new pharmaceutical agents (Figure 4.20).
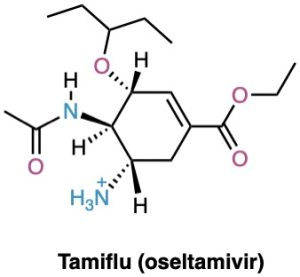
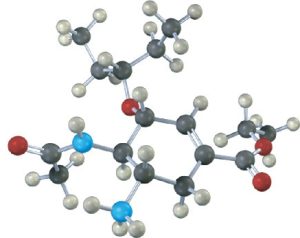
Figure 4.20 The structure of Tamiflu (oseltamivir), an antiviral agent active against type A influenza, along with a molecular model of its minimum-energy conformation as calculated by molecular mechanics.
