
From the description thus far, you might expect all 1H nuclei in a molecule to absorb energy at the same frequency and all 13C nuclei to absorb at the same frequency. If so, we would observe only a single NMR absorption signal in the 1H or 13C spectrum of a molecule, a situation that would be of little use. In fact, the absorption frequency is not the same for all 1H or all 13C nuclei.
All nuclei in molecules are surrounded by electrons. When an external magnetic field is applied to a molecule, the electrons moving around nuclei set up tiny local magnetic fields of their own. These local magnetic fields act in opposition to the applied field so that the effective field actually felt by the nucleus is a bit weaker than the applied field.
𝐵effective = 𝐵applied = 𝐵local
In describing this effect of local fields, we say that nuclei experience shielding from the full effect of the applied field by the surrounding electrons. Because each chemically distinct nucleus in a molecule is in a slightly different electronic environment, each nucleus is shielded to a slightly different extent and the effective magnetic field felt by each is slightly different. These tiny differences in the effective magnetic fields experienced by different nuclei can be detected, and we thus see a distinct NMR signal for each chemically distinct 13C or 1H nucleus in a molecule. As a result, an NMR spectrum effectively maps the carbon– hydrogen framework of an organic molecule. With practice, it’s possible to read this map and derive structural information.
Figure 13.4 shows both the 1H and the 13C NMR spectra of methyl acetate, CH3CO2CH3. The horizontal axis shows the effective field strength felt by the nuclei, and the vertical axis indicates the intensity of absorption of rf energy. Each peak in the NMR spectrum corresponds to a chemically distinct 1H or 13C nucleus in the molecule. Note that NMR spectra are formatted with the zero absorption line at the bottom, whereas IR spectra are formatted with the zero absorption line at the top; Section 12.5. Note also that 1H and 13C spectra can’t be observed simultaneously on the same spectrometer because different amounts of energy are required to spin-flip the different kinds of nuclei. The two spectra must be recorded separately.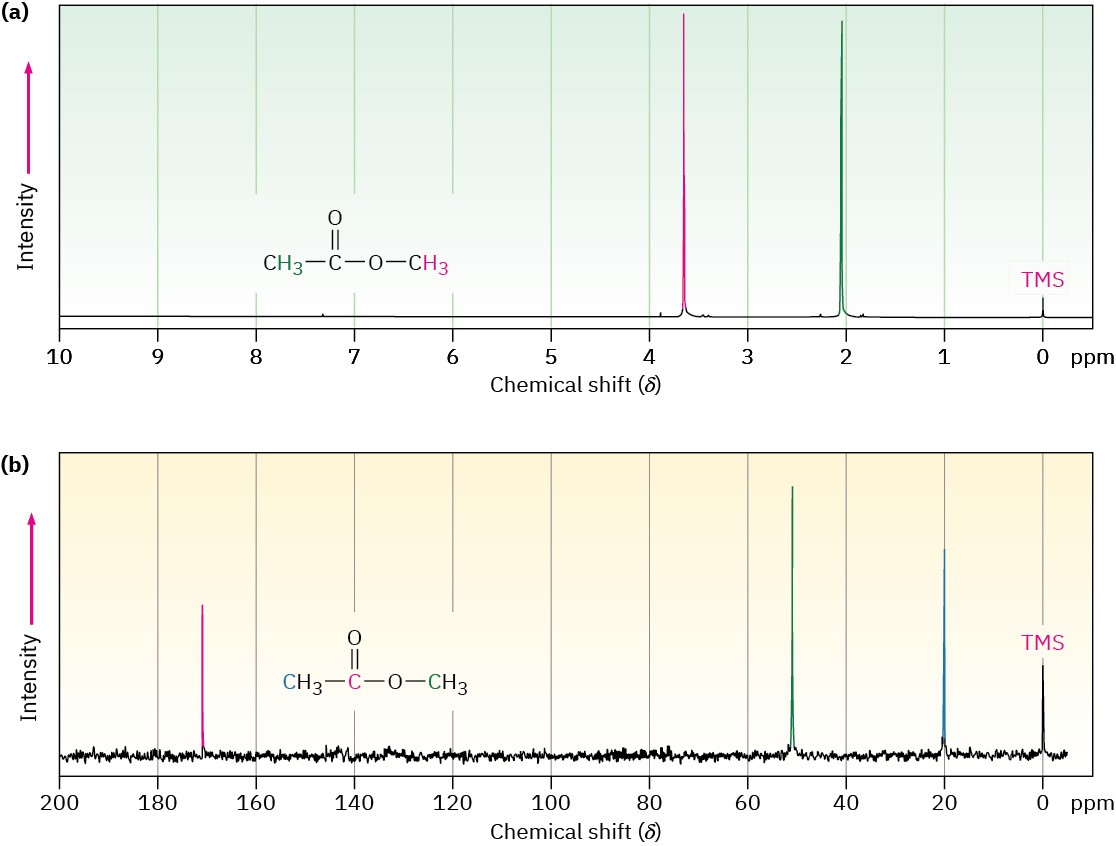
Figure 13.4 (a) The 1H NMR spectrum and (b) the proton-decoupled 13C NMR spectrum of methyl acetate, CH3CO2CH3. The small peak labeled “TMS” at the far right of each spectrum is a calibration peak, as explained in the next section.
The 13C NMR spectrum of methyl acetate in Figure 13.4b shows three peaks, one for each of the three chemically distinct carbon atoms in the molecule. The 1H NMR spectrum in Figure 13.4a shows only two peaks, however, even though methyl acetate has six hydrogens. One peak is due to the CH3C═O hydrogens, and the other to the −OCH3 hydrogens. Because the three hydrogens in each methyl group have the same electronic environment, they are shielded to the same extent and are said to be equivalent. Chemically equivalent nuclei always show the same absorption. The two methyl groups themselves, however, are not equivalent, so the two sets of hydrogens absorb at different positions.
The operation of a basic NMR spectrometer is illustrated in Figure 13.5. An organic sample is dissolved in a suitable solvent (usually deuteriochloroform, CDCl3, which has no hydrogens) and placed in a thin glass tube between the poles of a magnet. The strong magnetic field causes the 1H and 13C nuclei in the molecule to align in one of the two possible orientations, and the sample is irradiated with rf energy. If the frequency of the rf irradiation is held constant and the strength of the applied magnetic field is varied, each nucleus comes into resonance at a slightly different field strength. A sensitive detector monitors the absorption of rf energy, and its electronic signal is then amplified and displayed as a peak.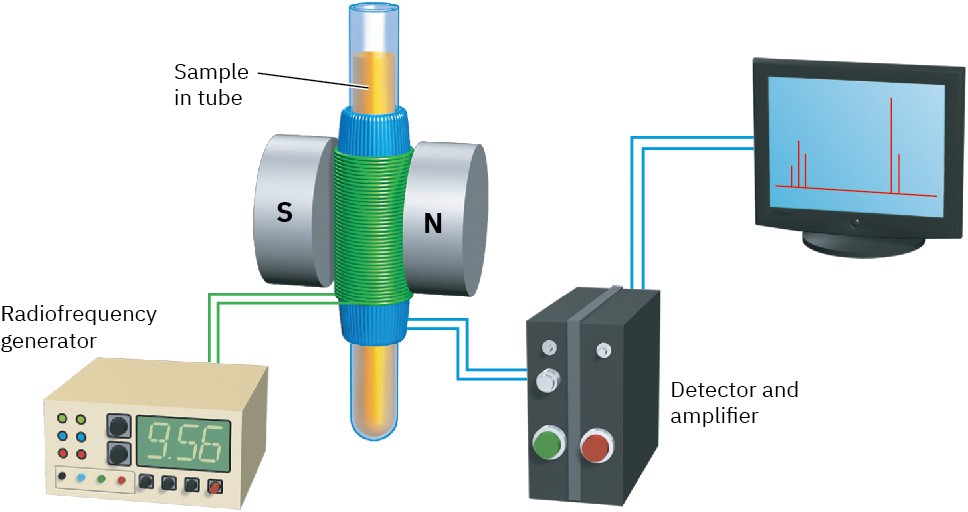
Figure 13.5 Schematic operation of a basic NMR spectrometer. A thin glass tube containing the sample solution is placed between the poles of a strong magnet and irradiated with rf energy.
NMR spectroscopy differs from IR spectroscopy (Section 12.6–Section 12.8) in that the timescales of the two techniques are quite different. The absorption of infrared energy by a molecule giving rise to a change in vibrational amplitude is an essentially instantaneous process (about 10–13 s), but the NMR process is much slower (about 10–3 s). This difference in timescales between IR and NMR spectroscopy is analogous to the difference between cameras operating at very fast and very slow shutter speeds. The fast camera (IR) takes an instantaneous picture and freezes the action. If two rapidly interconverting species are present, IR spectroscopy records the spectrum of both. The slow camera (NMR), however, takes a blurred, time-averaged picture. If two species interconverting faster than 103 times per second are present in a sample, NMR records only a single, averaged spectrum, rather than separate spectra of the two discrete species.
Because of this blurring effect, NMR spectroscopy can be used to measure the rates and activation energies of very fast chemical processes. In cyclohexane, for example, a ring-flip (Section 4.6) occurs so rapidly at room temperature that axial and equatorial hydrogens can’t be distinguished by NMR; only a single, averaged 1H NMR absorption is seen for cyclohexane at 25 °C. At –90 °C, however, the ring-flip is slowed down enough that two absorption peaks are visible, one for the six axial hydrogens and one for the six equatorial hydrogens. Knowing the temperature and the rate at which signal blurring begins to occur, it’s possible to calculate that the activation energy for the cyclohexane ring-flip is 45 kJ/mol.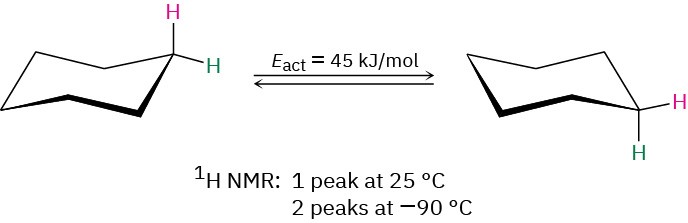
Problem 13-3
2-Chloropropene shows signals for three kinds of protons in its 1H NMR spectrum. Explain.
