
13 Chapter 13 – Structure Determination: Nuclear Magnetic Resonance Spectroscopy
| 13.1 | [latex]E = \frac{1.20×10^{-4} kJ/mol}{λ(in{\,}m)}[/latex] |
| [latex]λ = \frac{c}{v} = \frac{3.0×10^8 m/s}{v}; v = 187{\,}MHz = 1.87 × 10^8{\,}Hz[/latex] | |
| [latex]λ = \frac{3.0×10^8 m/s}{1.87×10^8} = 1.60 m[/latex] | |
| [latex]E = \frac{1.20×10^{-4} kJ/mol}{1.60} = 7.5×10^{-5} kJ/mol[/latex] | |
| Compare this value with E = 8.0 × 10–5 kJ/mol for 1H (given in the text). It takes slightly less energy to spin-flip a 19F nucleus than to spin-flip a 1H nucleus. |
| 13.2 | [latex]λ = \frac{c}{v} = \frac{3.0×10^8 m/s}{v}; v = 300{\,}MHz = 3 × 10^8{\,}Hz[/latex] |
| [latex]λ = \frac{3.0×10^8 m/s}{3×10^8} = 1.0 m[/latex] | |
| [latex]E = \frac{1.20×10^{-4} kJ/mol}{1.0} = 1.20×10^{-4} kJ/mol[/latex] | |
| Increasing the spectrometer frequency from 200 MHz to 300 MHz increases the amount of energy needed for resonance. |
| 13.3 | 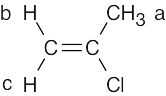
2-Chloropropene has three kinds of protons. Protons b and c differ because one is cis to the chlorine and the other is trans. |
| 13.4 | [latex]δ = \frac{Observed{\,}chemical{\,}shift{\,}(in{\,}Hz)}{200 MHz}[/latex] | |
| (a) | [latex]δ = \frac{1454 Hz}{200 MHz}[/latex] = δ 7.27 for CHCl3 | |
| (b) | [latex]δ = \frac{610 Hz}{200 MHz}[/latex] = δ 3.05 for CH3Cl | |
| (c) | [latex]δ = \frac{693 Hz}{200 MHz}[/latex] = δ 3.46 for CH3OH | |
| (d) | [latex]δ = \frac{1060 Hz}{200 MHz}[/latex] = δ 5.30 for CH2Cl2 | |
| 13.5 | (a) | [latex]δ = \frac{Observed{\,}chemical{\,}shift{\,}(number{\,}Hz{\,}away{\,}from{\,}TMS)}{Spectrometer{\,}frequency{\,}in{\,}MHz}[/latex]
Units of δ are parts per million. In this problem, δ = 2.1 ppm [latex]2.1{\,}ppm = \frac{Observed{\,}chemical{\,}shift}{200{\,}(MHz)}[/latex] 420 Hz = Observed chemical shift |
| (b) | If the 1H NMR spectrum of acetone were recorded at 500 MHz, the position of absorption would still be 2.1 δ because measurements given in ppm or δ units are independent of the operating frequency of the NMR spectrometer. | |
| (c) | [latex]2.1{\,}δ = \frac{Observed{\,}chemical{\,}shift}{500{\,}(MHz)}[/latex]; Observed chemical shift = 1050 Hz |
| 13.6 | (a) |
Four resonance lines are observed because of symmetry. |
| (b) | 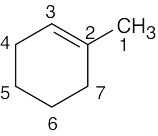 1-Methylcyclohexene Seven lines are seen because no two carbons are equivalent |
|
| (c) | 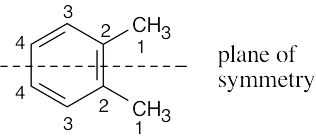
1,2-Dimethylbenzene Four resonance lines are seen. |
|
| (d) | 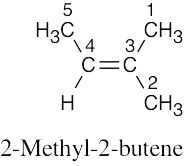
Five resonance lines are observed. Carbons 1 and 2 are nonequivalent because of the double bond stereo-chemistry. |
|
| (e) | 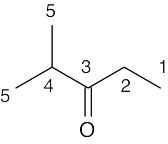
Five resonance lines are seen. |
|
| (f) | 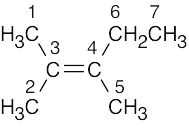
Seven resonance lines are seen. |
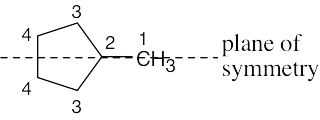
| 13.7 | Many other structures can be drawn. | |
| (a) | 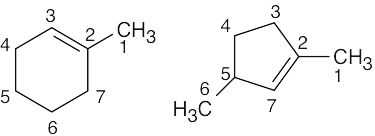 |
|
| (b) | 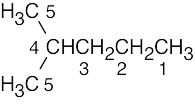
Two of the 6 carbons are equivalent. |
|
| (c) | 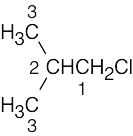
Two of the 4 carbons are equivalent. |
|
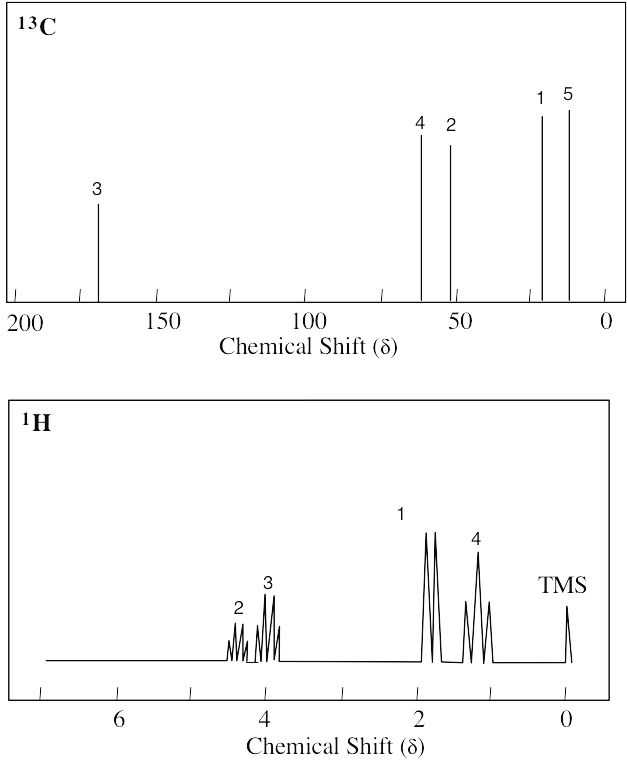
| 13.8 | Methyl propanoate has 4 unique carbons, and each one absorbs in a specific region of the 13C spectrum. The absorption (4) has the lowest value of δ and occurs in the –CH3 region of the 13C spectrum. Absorption (3) occurs in the –CH2– region. The methyl group (1) is next to an electronegative atom and absorbs downfield from the other two absorptions. The carbonyl carbon (2) absorbs the farthest downfield. | |||||||||||
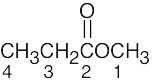 |
|
|||||||||||
| 13.9 |
The top spectrum shows all eight 13C NMR peaks. The middle spectrum (DEPT-90) shows only peaks due to CH carbons. From the DEPT-90 spectrum, the absorption at 124 δ can be assigned to the vinyl carbon (5), and the absorption at 68 δ can be assigned to the –OH carbon (2). The DEPT-135 spectrum shows all but the quaternary carbon (6), which appears in the top spectrum at 132 δ. The top half of the DEPT-135 spectrum shows absorptions due to CH3 carbons and CH carbons (which we have already identified). The 3 remaining peaks on the top of the DEPT-135 spectrum are due to methyl groups. Although we haven’t learned enough to distinguish among these peaks, the peak at 23 δ is due to carbon (1). The other two peaks arise from carbons (7) and (8) (18 δ, 26 δ). The bottom half of the DEPT-135 shows the two CH2 carbons. Carbon (3) absorbs at 39 δ (negative), and carbon (4) absorbs at 24 δ (negative). |
||||||||||||||||||
 |
|
||||||||||||||||||
| 13.10 | Identify the carbons as CH3, CH2, CH or quaternary, and use Figure 13.8 to find approximate values for chemical shifts. (When an actual spectrum is given, it is easier to assign the carbons to the chemical shifts.) Remember: DEPT-90 spectra identify CH carbons, and DEPT-135 spectra identify CH3 carbons (positive peaks), CH carbons (positive peaks already identified), and CH2 carbons (negative peaks). Quaternary carbons are identified in the broadband-decoupled spectrum, in which all peaks appear. | |||||||||||||||||||||||||||||||||
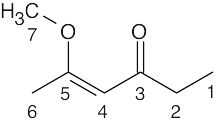 |
|
|||||||||||||||||||||||||||||||||
| 13.11 |
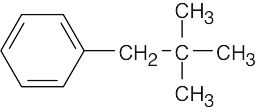
Always start this type of problem by calculating the degree of unsaturation of the unknown compound. C11H16 has 4 degrees of unsaturation. Since the unknown hydrocarbon is aromatic, a benzene ring accounts for all four degrees of unsaturation. Next, look for elements of symmetry. Although the molecular formula indicates 11 carbons, only 7 peaks appear in the 13C NMR spectrum, indicating a plane of symmetry. Four of the 7 peaks are due to aromatic carbons, indicating a benzene ring that is probably monosubstituted. (Prove to yourself that a monosubstituted benzene ring has 4 different kinds of carbons). The DEPT-90 spectrum shows that 3 of the kinds of carbons in the aromatic ring are CH carbons. The positive peaks in the DEPT-135 spectrum include these three peaks, along with the peak at 29.5 δ, which is due to a CH3 carbon. The negative peak in the DEPT- 135 spectrum is due to a CH2 carbon. Two peaks remain unidentified and are thus quaternary carbons; one of them is aromatic. At this point, the unknown structure is a monosubstituted benzene ring with a substituent that contains CH2, C, and CH3 carbons. A structure for the unknown compound that satisfies all data:
|
| 13.12 | 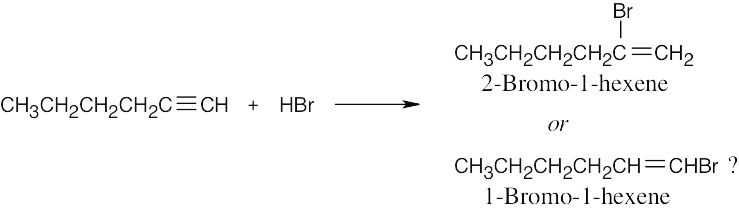
The two possible products are easy to distinguish by using 13C NMR. 2-Bromo-1-hexene, the actual product formed, shows no peaks in its DEPT-90 13C NMR spectrum because it has no CH carbons. The other possible product, 1-bromo-1-hexene, shows 2 peaks in its DEPT-90 spectrum. |
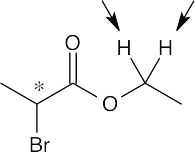
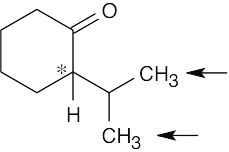
| 13.13 | First, check for protons that are unrelated (none appear in this problem). Next, look for molecules that already have chirality centers. Replacement of a –CH2– proton by X in (d) and (e) produces a second chirality center, and the two possible replacement products are diastereomers. Thus, the indicated protons in (d) and (e) are diastereotopic.
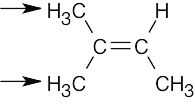
Finally, for the other molecules, mentally replace each of the two hydrogens in the indicated set with X, a different group. In (a), the resulting products are enantiomers, and the protons are enantiotopic. Replacement of the protons in (b) produces two chirality centers (the carbon bearing the hydroxyl group is now chiral) and the indicated protons are diastereotopic. Replacement of one of the methyl protons in each of the groups in (c) produces a pair of double-bond isomers that are diastereomers; these protons are diastereotopic. The protons in (f) are homotopic, producing only one signal. |
|
| (a) | enantiotopic
|
|
| (b) | diastereotopic
|
|
| (c) | diastereotopic
|
|
| (d) | Diastereotopic
|
|
| (e) | Diastereotopic
|
|
| (f) | Homotopic
|
|
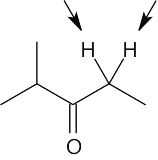
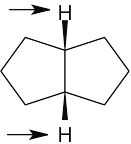
| 13.14 | Compound | Kinds of non-equivalent protons | |
| (a) | 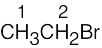 |
2 | |
| (b) | 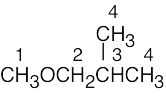 |
4 | |
| (c) | 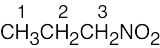 |
3 | |
| (d) | 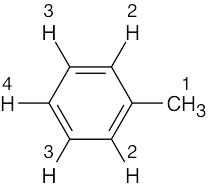 |
4 | |
| (e) |
|
5 | |
| (f) |
|
3 |
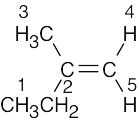
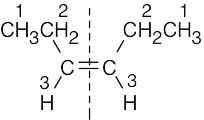
| 13.15 | 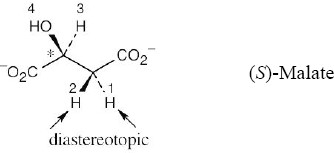
Because (S)-malate already has a chirality center (starred), the two protons next to it are diastereotopic and absorb at different values. The 1H NMR spectrum of (S)-malate has four absorptions. |
| 13.16 | Compound | δ | Kinds of non-equivalent protons | |
| (a) | C6H12 | 1.43 | secondary alkyl | |
| (b) | CH3COCH3 | 2.17 | methyl ketone | |
| (c) | C6H6 | 7.37 | aromatic | |
| (d) | CH2Cl2 | 5.30 | protons adjacent to two halogens | |
| (e) | OHCCHO | 9.70 | aldehyde | |
| (f) | (CH3)3N | 2.12 | methyl protons adjacent to nitrogen |
| 13.17 | 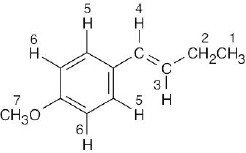 |
|
||||||||||||||||||||||||
| This compound has seven different kinds of protons. Notice that the two protons labeled 5 are equivalent, as are the two protons labeled 6 because of rotation around the bond joining the aromatic ring and the alkenyl side chain. | ||||||||||||||||||||||||||
| 13.18 | 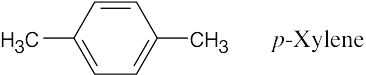
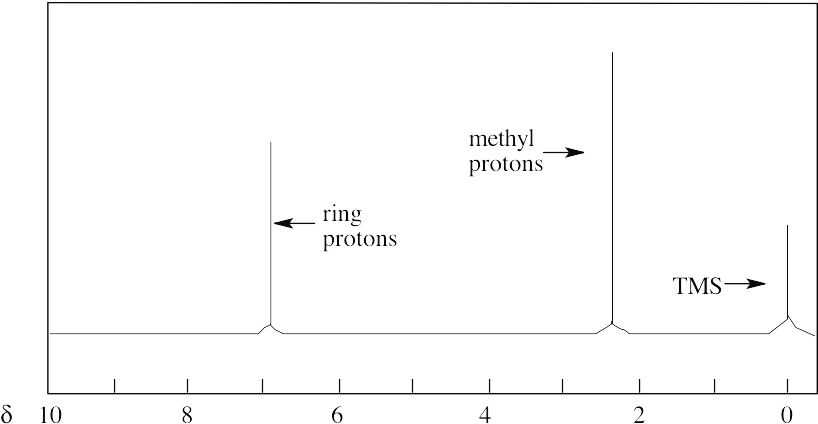
There are two absorptions in the 1H NMR spectrum of p–xylene. The four ring protons absorb at 7.05 δ, and the six methyl-group protons absorb at 2.23 δ. The peak ratio of methyl protons:ring protons is 3:2.
|
| 13.19 | Compound | Protons | Number of Adjacent Protons | Splitting | |
| (a) | 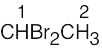 |
1 | 3 | quartet | |
| 2 | 1 | doublet | |||
| (b) | 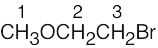 |
1 | 0 | singlet | |
| 2 | 2 | triplet | |||
| 3 | 2 | triplet | |||
| (c) | 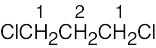 |
1 | 2 | triplet | |
| 2 | 4 | quintet | |||
| (d) | 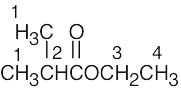 |
1 | 1 | doublet | |
| 2 | 6 | septet | |||
| 3 | 3 | quartet | |||
| 4 | 2 | triplet | |||
| (e) | 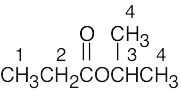 |
1 | 2 | triplet | |
| 2 | 3 | quartet | |||
| 3 | 6 | septet | |||
| 4 | 1 | doublet | |||
| (f) | 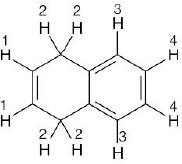 |
1 | 2 | triplet | |
| 2 | 1 | doublet | |||
| 3 | 1 | multiplet | |||
| 4 | 1 | multiplet | |||
| The splitting patterns for protons 3 and 4 are complex and are not explained in the text. | |||||
| 13.20 | Calculate the degree of unsaturation, and note the number of peaks to see if symmetry is present. | |||||||
| (a) | This compound has no degrees of saturation and only one kind of hydrogen. The only possible structure is CH3OCH3. | |||||||
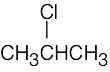
| (b) | Again, this compound has no degrees of unsaturation and has two kinds of hydrogens. The compound is 2-chloropropane.
|
|||||||
| (c) | This compound, with no degrees of unsaturation, has two different kinds of hydrogen, each of which has two neighboring hydrogens. | |||||||
| (d) | C4H8O2; one degree of unsaturation and 3 different kinds of hydrogen.
|
|||||||
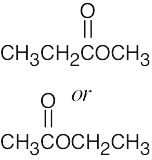
| 13.21 | The molecular formula (C4H10O) indicates that the compound has no multiple bonds or rings. The 1H NMR spectrum shows two signals, corresponding to two types of hydrogens in the ratio 1.50:1.00, or 3:2. Since the unknown contains 10 hydrogens, four protons are of one type and six are of the other type.
The upfield signal at 1.22 δ is due to saturated primary protons. The downfield signal at 3.49 δ is due to protons on carbon adjacent to an electronegative atom – in this case, oxygen. The signal at 1.23 δ is a triplet, indicating two neighboring protons. The signal at 3.49 δ is a quartet, indicating three neighboring protons. This splitting pattern is characteristic of an ethyl group. The compound is diethyl ether, CH3CH2OCH2CH3. |
| 13.22 | 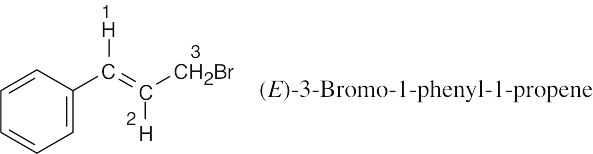
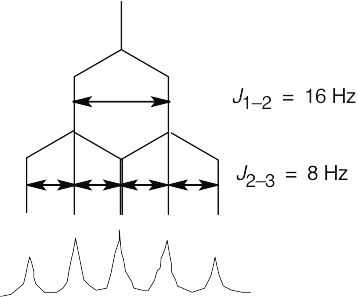
Coupling of the C2 proton to the Cl vinylic proton occurs with J = 16 Hz and causes the signal of the C2 proton to be split into a doublet. The C2 proton is also coupled to the two C3 protons with J = 8 Hz. This splitting causes each leg of the C2 proton doublet to be split into a triplet, producing six lines in all. Because of the size of the coupling constants, two of the lines coincide, and a five-line multiplet is observed.
|
| 13.23 | 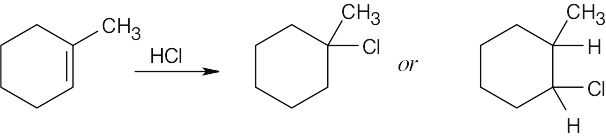
Focus on the 1H NMR methyl group absorption. In the left product, the methyl group signal is unsplit; in the right product, it appears as a doublet. In addition, the right product shows a downfield absorption in the 2.5 δ – 4.0 δ region due to the proton bonded to a carbon that is also bonded to an electronegative atom. If you were to take the 1H NMR spectrum of the reaction product, you would find an unsplit methyl group, and you could conclude that the product was 1-chloro-1-methylcyclohexane. |
Additional Problems
Visualizing Chemistry
| 13.24 | (a) | 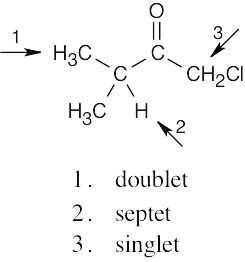 |
| (b) | 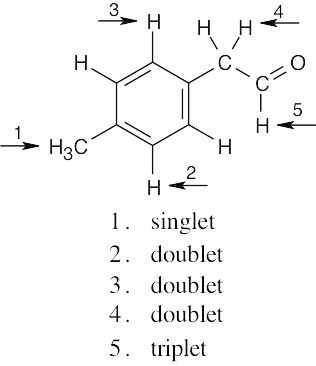 |
| 13.25 | None of the hydrogens or carbons are equivalent.
|

| 13.26 | The compound has 5 different types of carbons and 4 different types of hydrogens.
|
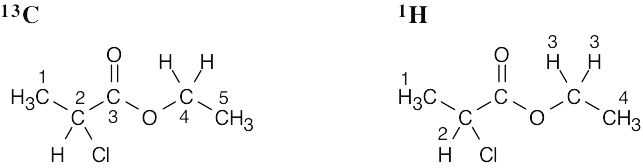
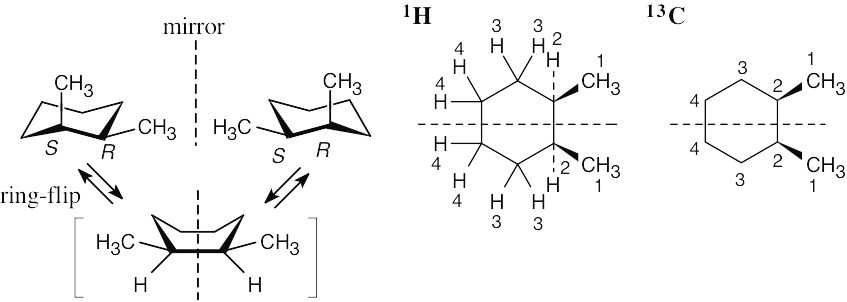
| 13.27 | If you assign R, S configurations to the two carbons bonded to the methyl group, it is apparent that cis-1,2-dimethylcyclohexane is a meso compound. When the cyclohexane ring undergoes a ring-flip, the ring passes through an intermediate that has a plane of symmetry. Both the 13C NMR spectrum and the 1H NMR spectrum show 4 peaks.
|
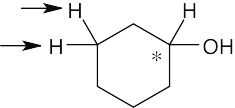
| 13.28 | (a) | Because cysteine has a chirality center, the indicated protons are diastereotopic. |
| (b) | Imagine replacing first one, then the other, of the indicated protons with a substituent X. The two resulting compounds would be enantiomers. The protons are thus enantiotopic. | |
| (a) | 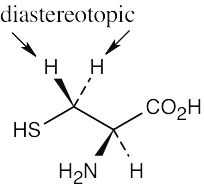 |
|
| (b) | 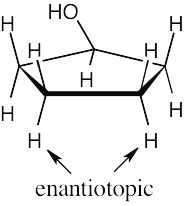 |
Chemical Shifts and NMR Spectroscopy
| 13.29 | [latex]δ = \frac{Observed{\,}chemical{\,}shift{\,}(in{\,}Hz)}{200{\,}MHz}[/latex] | |
| (a) | 2.18 δ | |
| (b) | 4.78 δ | |
| (c) | 7.52 δ | |
| 13.30 | δ × 300 MHz = Observed chemical shift (in Hz) | |
| (a) | 30 Hz | |
| (b) | 1035 Hz | |
| (c) | 1890 Hz | |
| (d) | 2310 Hz | |
| 13.31 | (a) | Since the symbol “δ” indicates ppm downfield from TMS, chloroform absorbs at 7.3 ppm. |
| (b) | [latex]δ = \frac{Observed{\,}chemical{\,}shift{\,}(in{\,}Hz)}{Spectrophotometer{\,}frequency{\,}in{\,}MHz}[/latex] | |
| [latex]7.3 ppm = \frac{chemical{\,}shift{\,}(in{\,}Hz)}{360{\,}(MHz)}[/latex]; 7.3 ppm × 360 MHz = chemical shift | ||
| 2600 Hz = Observed chemical shift | ||
| (c) | The value of δ is still 7.3 because the chemical shift measured in δ is independent of the operating frequency of the spectrometer. |
| 13.32 | 13C NMR absorptions occur over a range of 250 ppm, while 1H NMR absorptions generally occur over a range of only 10 ppm. The spread of peaks in 13C NMR is therefore much greater, so accidental overlap is less likely. In addition, normal 13C NMR spectra are uncomplicated by spin–spin splitting, and the total number of lines is smaller. |
| 13.33 | A nucleus that absorbs at 6.50 δ is less shielded than a nucleus that absorbs at 3.20 δ and thus requires a weaker applied field to come into resonance. A shielded nucleus feels a smaller effective field, and a stronger applied field is needed to bring it into resonance. |
1NMR Spectroscopy
| 13.34 | Compound | Kinds of non-equivalent protons | ||
| (a) | 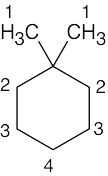 |
4 | ||
| (b) | 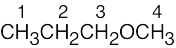 |
4 | ||
| (c) | 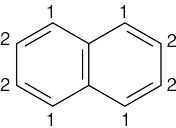 |
2 | ||
| (d) | 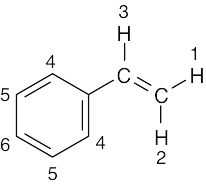 |
6 | ||
| (e) | 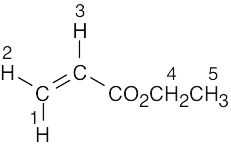 |
5 | ||
| 13.35 | Lowest Chemical Shift ⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯→ Highest Chemical Shift |
||||
| CH4 < | Cyclohexane < | CH3COCH3 < | CH2, H2C=CH2 < | Benzene | |
| 0.23 | 1.43 | 2.17 | 5.30, 5.33 | 7.37 | |
| 13.36 | (a) | 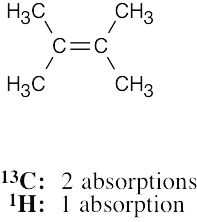 |
| (b) | 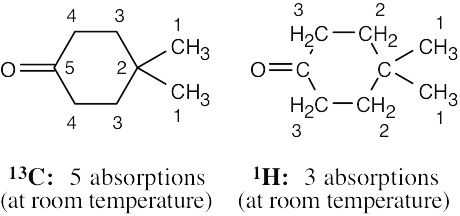 |
|
| (c) | 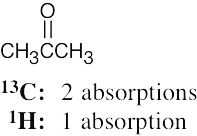 |
|
| (d) | 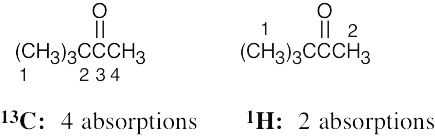 |
|
| (e) | 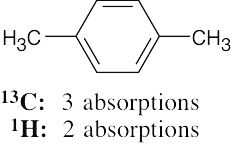 |
|
| (f) | 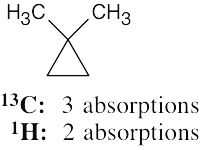 |
| 13.37 | (a) | (CH3)4C |
| (b) | 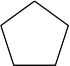 |
|
| (c) | 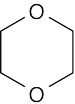 |
| 13.38 | Compound | Number of peaks | Peak assignment | Splitting Pattern | |
| (a) | 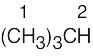 |
2 | 1 | doublet (9H) | |
| 2 | multiplet (1H) (dectet) | ||||
| (b) | 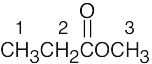 |
3 | 1 | triplet (3H) | |
| 2 | quartet (2H) | ||||
| 3 | singlet (3H) | ||||
| (c) | 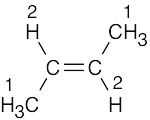 |
2 | 1 | doublet (6H) | |
| 2 | quartet (2H) |
| 13.39 | Peak assignment | Splitting Pattern | |
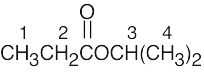 |
1 | triplet (3H) | |
| 2 | quartet (2H) | ||
| 3 | septet (1H) | ||
| 4 | doublet (6H) |
| 13.40 | (a) | enantiotopic
|
| (b) | diastereotopic
|
|
| (c) | diastereotopic
|
|
|
Refer to Problem 13.13 for help. The protons in (c) are diastereotopic because the molecules that result from replacement of the indicated hydrogens are diastereomers (prove it to yourself with models). |
||
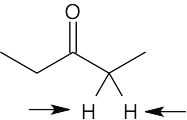
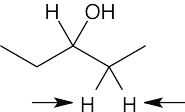
| 13.41 | (a) | homotopic
|
| (b) | enantiotopic
|
|
| (c) | diastereotopic
|
| 13.42 | Use of 13C NMR to distinguish between the two isomers has been described in the text in Section 13.7. 1H NMR can also be useful.
Isomer A has only four kinds of protons because of symmetry. Its vinylic proton absorption (4.5–6.5 δ) represents two hydrogens. Isomer B contains six different kinds of protons. Its 1H NMR shows an unsplit methyl group signal and one vinylic proton signal of relative area 1. These differences make it possible to distinguish between A and B. |

| 13.43 | First, check each isomer for structural differences that are recognizable in the 1H NMR spectrum. If it’s not possible to pick out distinguishing features immediately, it may be necessary to sketch an approximate spectrum of each isomer for comparison. | |||||||
| (a) | CH3CH=CHCH2CH3 has two vinylic protons with chemical shifts at 5.4–5.5 δ. Because ethylcyclopropane shows no signal in this region, it should be easy to distinguish one isomer from the other. | |||||||
| (b) | CH3CH2OCH2CH3 has two kinds of protons, and its 1H NMR spectrum consists of two peaks — a triplet and a quartet. CH3OCH2CH2CH3 has four different types of protons, and its spectrum is more complex. In particular, the methyl group bonded to oxygen shows an unsplit singlet absorption. | |||||||
| (c) | Each compound shows three peaks in its 1H NMR spectrum. The ester, however, shows a downfield absorption due to the –CH2– hydrogens next to oxygen. No comparable peak shows in the spectrum of the ketone.
|
|||||||
| (d) | Each isomer contains four different kinds of protons — two kinds of methyl protons and two kinds of vinylic protons. For the first isomer, the methyl peaks are both singlets, whereas for the second isomer, one peak is a singlet and one is a doublet. | |||||||
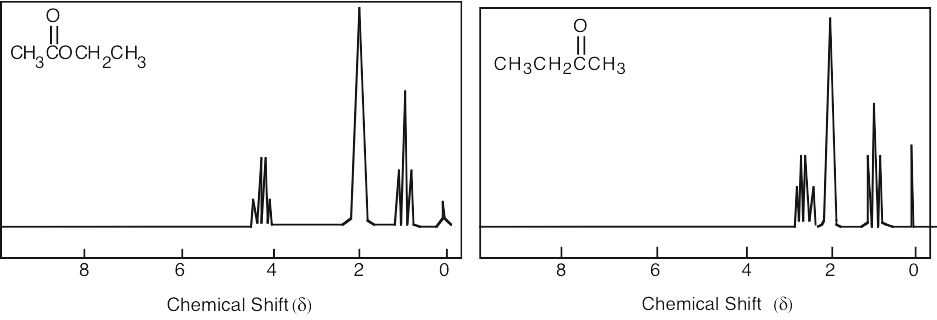
| 13.44 | (a) | 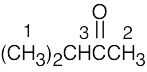
1 = 0.95 δ (isopropyl group) 2 = 2.10 δ (methyl ketone) 3 = 2.43 δ (isopropyl group) |
| (b) | 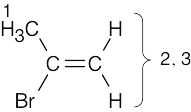
1 = 2.32 δ (methyl group attached to double bond) 2,3 = 5.35 δ, 5.54 δ (vinylic H) |
| 13.45 | (a) | 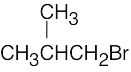 |
| (b) | 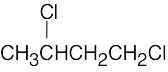 |
13C NMR Spectroscopy
| 13.46 |
|
| 13.47-13.48 | Compound | Number of 13C Absorptions | Carbons Showing Peaks in DEPT-135 13C NMR Spectrum | |||
| Positive Peaks | Negative Peaks | No Peaks | ||||
| (a) | 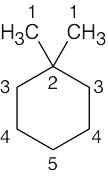 |
5 | carbon 1 | carbons 3, 4, 5 | carbon 2 | |
| (b) | 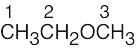 |
3 | carbons 1, 3 | carbon 2 | ||
| (c) | 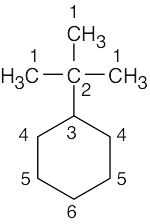 |
6 | carbons 1, 3 | carbons 4, 5, 6 | carbon 2 | |
| (d) | 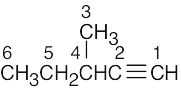 |
6 | carbons 1, 3, 4, 6 | carbon 5 | carbon 2 | |
| (e) | 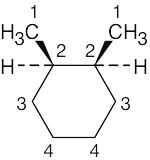 |
4 | carbons 1, 2 | carbons 3, 4 | ||
| (f) | 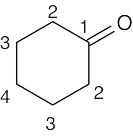 |
4 | carbons 2, 3, 4 | carbon 1 | ||
| 13.49 | Either 1H NMR or 13C NMR can be used to distinguish among these isomers. In either case, it is first necessary to find the number of different kinds of protons or carbon atoms.
13C NMR is the simplest method for identifying these compounds because each isomer differs in the number of absorptions in its 13C NMR spectrum. 1H NMR can also be used to distinguish among the isomers because the two isomers that show two 1H NMR peaks differ in their splitting patterns. |

| 13.50 |
Number of peaks |
Distinguishing Absorptions | ||
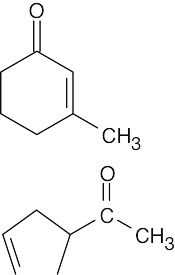 |
13C | 7 | Two vinylic peaks | |
| 1H | 5 | Unsplit vinylic peak, relative area 1 | ||
| 13C | 5 | One vinylic peak | ||
| 1H | 4 | Split vinylic peak, relative area 2 | ||
| The two isomers have different numbers of peaks in both 1H NMR and 13C NMR. In addition, the distinguishing absorptions in the vinylic region of both the 1H and 13C spectra make it possible to identify each isomer by its NMR spectrum.
The ketone IR absorption of 3-methyl-2-cyclohexenone occurs near 1690 cm–1 because the double bond is one bond away from the ketone group. The ketone IR absorption of 3-cyclopentenyl methyl ketone occurs near 1715 cm–1, the usual position for ketone absorption. |
||||
| 13.51 |
|

General Problems
| 13.52 | (a), (b) | C3H6O contains one double bond or ring. Possible structures for C3H6O include:
|
| (b) | Saturated ketones absorb at 1715 cm–1 in the infrared. Only the last two compounds above show an infrared absorption in this region. | |
| (c) | Because the aldehyde from part (b) has three different kinds of protons, its 1H NMR spectrum shows three peaks. The ketone, however, shows only one peak. Since the unknown compound of this problem shows only one 1H NMR absorption (in the methyl ketone region), it must be acetone. |
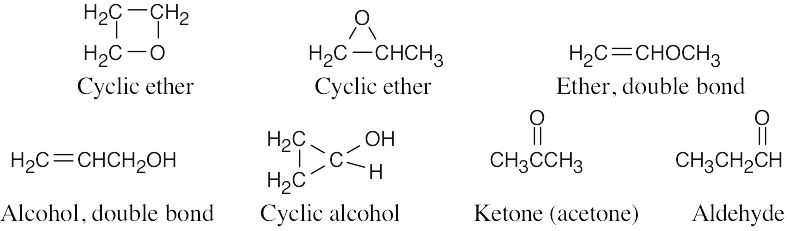
| 13.53 | The unknown compound has no degrees of unsaturation and has two different kinds of hydrogens. The unknown compound is BrCH2CH2CH2Br. |
| 13.54 | Possible structures for C4H7ClO2 (one degree of unsaturation) are CH3CH2CO2CH2Cl and ClCH2CO2CH2CH3. Chemical shift data can distinguish between them.
In A, the protons attached to the carbon bonded to both oxygen and chlorine (–OCH2Cl) are expected to absorb far downfield (5.0–6.0 δ). Because no signal is present in this region of the 1H NMR spectrum given, the unknown must be B. In addition, the quartet absorbing at 4.26 δ is typical of a CH2 group next to an electronegative atom and coupled with a methyl group. |

| 13.55 | (a) | 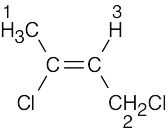
1 = 2.18 δ (allylic) 2 = 4.16 δ (H, Cl bonded to same C) 3 = 5.71 δ (vinylic) |
| (b) | 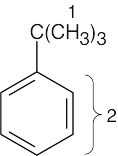
1 = 1.30 δ (saturated) 2 = 7.30 δ (aromatic) |
|
| (c) | 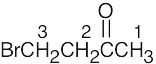
1 = 2.11 δ (next to C=O) 2 = 3.52 δ (next to C=O, Br) 3 = 4.40 δ (H, Br bonded to same C) The E isomer is also a satisfactory answer |
|
| (d) | 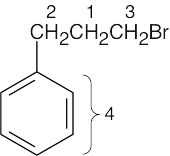
1 = 2.15 δ 2 = 2.75 δ (benzylic) 3 = 3.38 δ (H, Br bonded to same C) 4 = 7.22 δ (aromatic) |
|
|
In (b) and (d), the aromatic ring hydrogens coincidentally have the same chemical shift. |
| 13.56 | 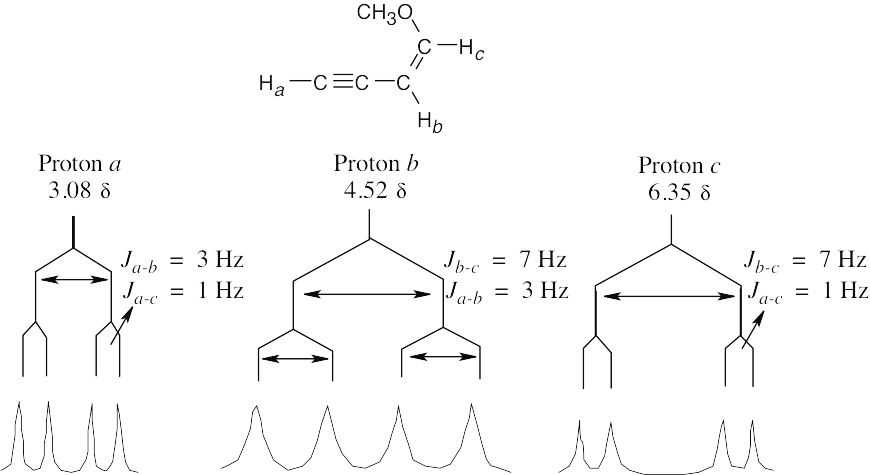 |
| 13.57 | Compound A (4 multiple bonds and/or rings) must be symmetrical because it exhibits only six peaks in its 13C NMR spectrum. Saturated carbons account for two of these peaks (δ = 15, 28 ppm), and unsaturated carbons account for the other four (δ = 119, 129, 131, 143 ppm).
1H NMR shows a triplet (3 H at 1.20 δ), and a quartet (2 H at 2.58 δ), indicating the presence of an ethyl group. The other signals (4 H at 7.07 δ, 7.39 δ are due to aromatic protons.
|
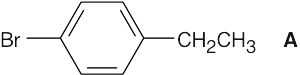
| 13.58 | (a) | 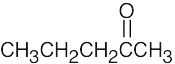 |
| (b) | 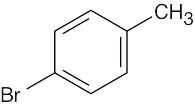 |
|
| (c) | 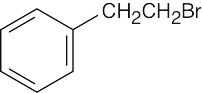 |
| 13.59 |
The peak in the mass spectrum at m/z = 84 is probably the molecular ion of the unknown compound and corresponds to a formula of C6H12 – one double bond or ring. The base peak, at m/z = 55, corresponds to the loss of an ethyl group. 13C NMR shows three different kinds of carbons and indicates a symmetrical hydrocarbon. The absorption at 132 δ is due to a vinylic carbon atom. A reasonable structure for the unknown is 3-hexene. The data do not distinguish between cis and trans isomers. CH3CH2CH=CHCH2CH3 3-Hexene |
| 13.60 | Compound A, a hydrocarbon having M+ = 96, has the formula C7H12, indicating two degrees of unsaturation. Because it reacts with BH3, Compound A contains a double bond. From the broadband decoupled 13C NMR spectrum, we can see that C7H12 is symmetrical, since it shows only five peaks.
The DEPT-135 spectrum of Compound A indicates three different CH2 carbons, one =CH2 carbon and one –C= carbon; the last two carbons are shown to be sp2-hybridized by their chemical shifts. In the DEPT-135 spectrum of Compound B, the absorptions due to double bond carbons have been replaced by a CH carbon and a CH2 carbon bonded to an electronegative group.
|
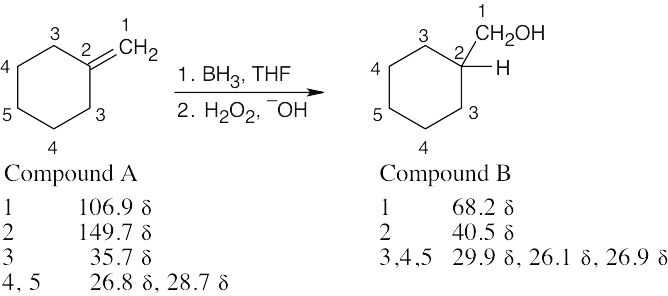
| 13.61 | The IR absorption indicates that C is an alcohol. From M+, we can arrive at a molecular formula of C5H10O, which indicates one degree of unsaturation. The broadband-decoupled spectrum shows five peaks; two are due to a double bond, and one is due to a carbon bonded to an electronegative atom (O).
The DEPT spectra show that C contains 4 CH2 carbons and one CH carbon, and that C has a monosubstituted double bond. HOCH2CH2CH2CH=CH2 is a likely structure for C. |
| 13.62 | Compound D is very similar to Compound C. The DEPT spectra make it possible to distinguish between the isomers. D has 2 CH carbons, one CH3 carbon, and 2 CH2 carbons, and, like C, has a monosubstituted double bond. The peak at 74.4 δ is due to a secondary alcohol.
|
| 13.63 | Compound E, C7H12O2, has two degrees of unsaturation and has two equivalent carbons because its broadband-decoupled spectrum shows only 6 peaks. Two carbons absorb in the vinylic region of the spectrum; because one is a CH carbon and the other is a CH2 carbon, E contains a monosubstituted double bond. The peak at 165.8 δ (not seen in the DEPT spectra) is due to a carbonyl group.
|

| 13.64 | 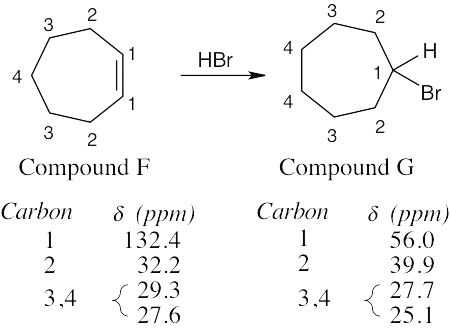 |
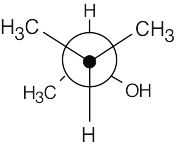
| 13.65 | Make a model of one enantiomer of 3-methyl-2-butanol and orient it as a staggered Newman projection along the C2-C3 bond. The S enantiomer is pictured.
Because of the chirality center at C2, the two methyl groups at the front of the projection are diastereotopic. Since the methyl groups aren’t equivalent, their carbons show slightly different signals in the 13C NMR. |
| 13.66 | Commercial 2,4-pentanediol is a mixture of three stereoisomers: (R,R), (S,S), and (R,S). The meso isomer shows three signals in its 13C NMR spectrum. Its diastereomers, the R,R and S,S enantiomeric pair, also show three signals, but two of these signals occur at different δ values from the meso isomer. This is expected, because diastereomers differ in physical and chemical properties. One resonance from the meso compound accidentally overlaps with one signal from the enantiomeric pair. |
| 13.67 | The product (M+= 88) has the formula C4H8O2. The IR absorption indicates that the product is an ester. The 1H NMR shows an ethyl group and an –OCH3 group.
|
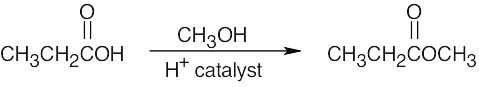
| 13.68 | The product is a methyl ketone.
|
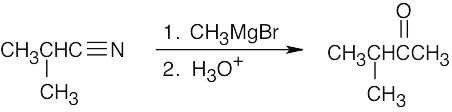
| 13.69 | Ethyl 2-nitropropanoate
|
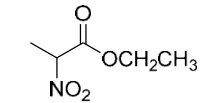
| 13.70 | 3-Methyl-3-buten-1-ol
|

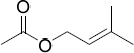
| 13.71 | Prenyl acetate (3-methyl-2-butenyl ethanoate)
|
This file is copyright 2023, Rice University. All Rights Reserved.
