
14 Chapter 14 – Conjugated Compounds and Ultraviolet Spectroscopy
Chapter 14 – Conjugated Compounds and Ultraviolet Spectroscopy
Solutions to Problems
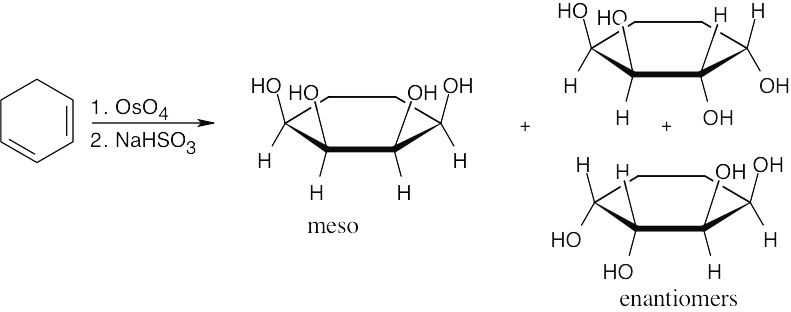
| 14.1 | We would expect ΔHhydrog = –126 + (–126) = –252 kJ/mol for allene if the heat of hydrogenation for each double bond were the same as that for an isolated double bond. The measured ΔHhydrog, –298 kJ/mol, is 46 kJ/mol more negative than the expected value. Thus, allene is higher in energy (less stable) than a nonconjugated diene, which in turn is less stable than a conjugateddiene. |
| 14.2 |
|
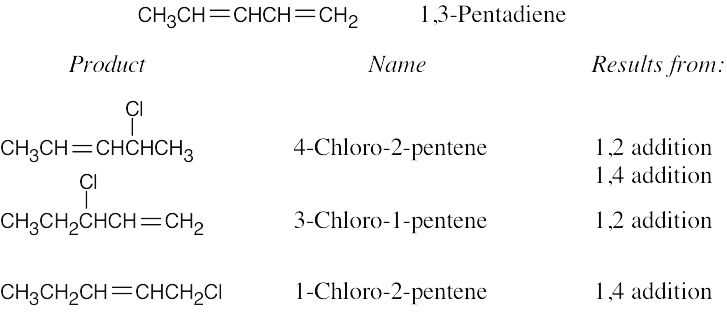
| 14.3 |
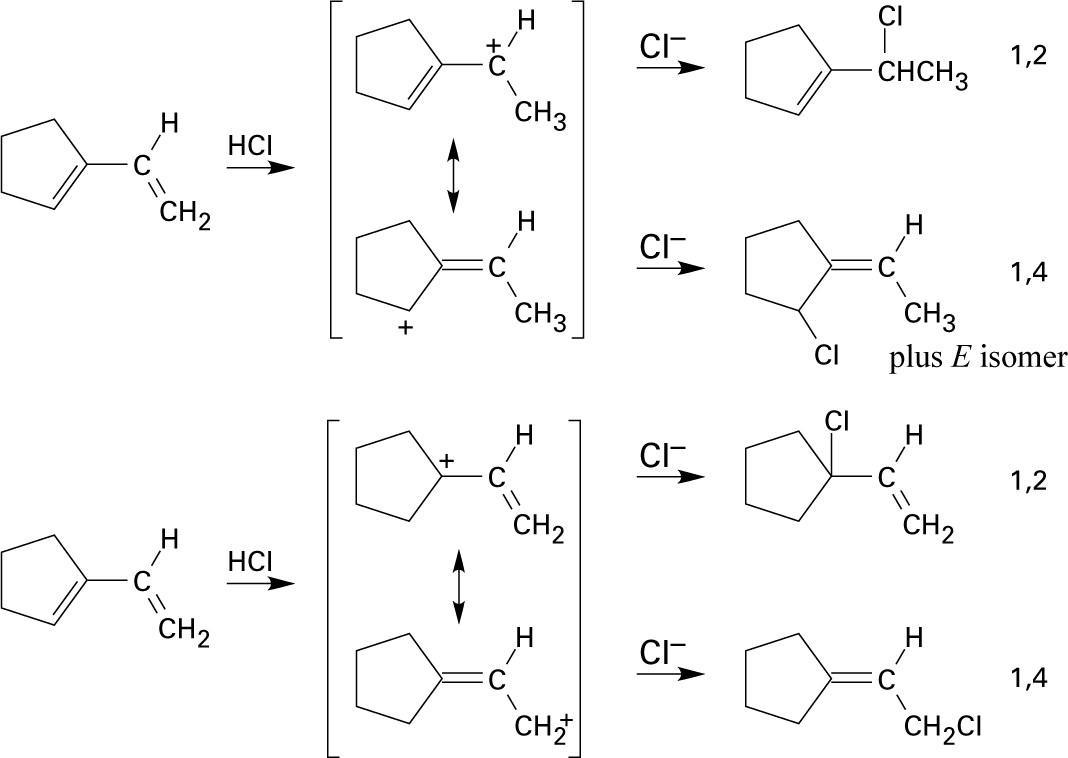
A and D, which are resonance-stabilized, are formed in preference to B and C, which are not. The positive charge of allylic carbocation A is delocalized over two secondary carbons, while the positive charge of carbocation D is delocalized over one secondary and one primary carbon. We therefore predict that carbocation A is the major intermediate formed, and that 4-chloro-2-pentene predominates. Note that this product results from both 1,2 and 1,4 addition. |
| 14.4 |
|
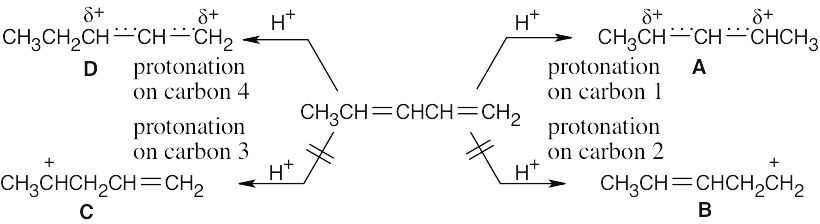
| 14.5 |
Allylic halides can undergo slow dissociation to form stabilized carbocations (SN1 reaction). Both 3-bromo-1-butene and 1-bromo-2-butene form the same allylic carbocation, pictured above, on dissociation. Addition of bromide ion to the allylic carbocation then occurs to form a mixture of bromobutenes. Since the reaction is run under equilibrium conditions, the thermodynamically more stable 1-bromo-2-butene predominates. |
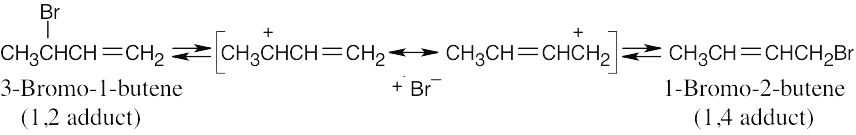
| 14.6 |
1,4 adducts are more stable than 1,2 adducts because disubstituted double bonds are more stable than monosubstituted double bonds (see Chapter 7). |
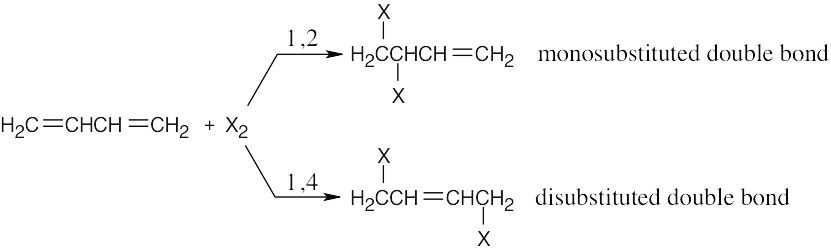
| 14.7 | Draw the reactants in an orientation that shows where the new bonds will form. Form the new bonds by connecting the two reactants, removing two double bonds, and relocating the remaining double bond so that it lies between carbon 2 and carbon 3 of the diene. The substituents on the dienophile retain their trans relationship in the product. The product is a racemic mixture.
|
| 14.8 | Good dienophiles have an electron-withdrawing group conjugated with a double bond.
Compounds (a) and (d) are good dienophiles because they have electron-withdrawing groups conjugated with a carbon–carbon double bond. Alkene (c) is a poor dienophile because it has no electron-withdrawing functional group. Compounds (b) and (e) are poor dienophiles because their electron-withdrawing groups are not conjugated with the double bond. |
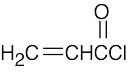
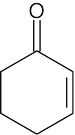
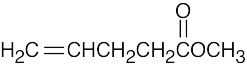
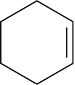
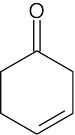
| 14.9 |
|
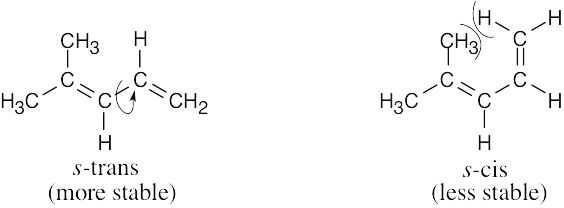
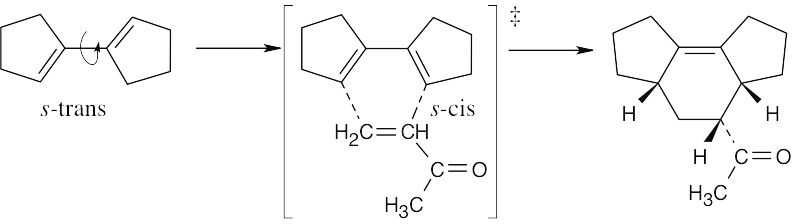
| 14.10 | Rotation of the diene to the s-cis conformation must occur in order for a reaction to take place.
|
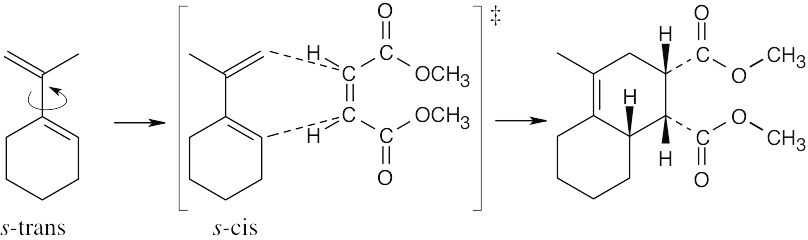
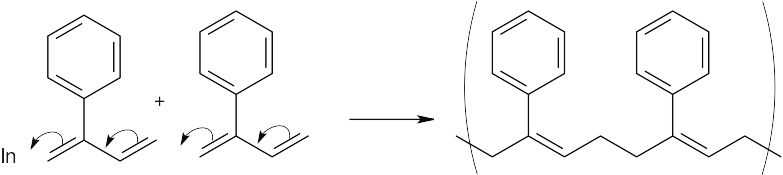
| 14.11 | The initiator may be either a radical or a cation. Diene polymerization is a 1,4 addition process that forms a polymer whose monomer units have a 4 carbon chain that contains a double bond every 4 bonds.
|
| 14.12 |
|
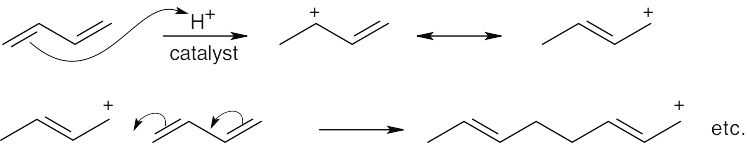
| 14.13 |
|


| 14.14 |
|

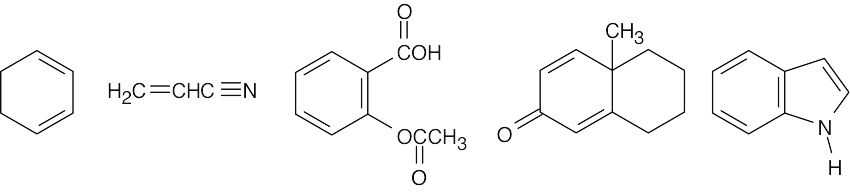
| 14.15 | All compounds having alternating single and multiple bonds should show ultraviolet absorption in the range 200–400 nm. Only compound (a) is not UV-active. All of the compounds pictured below (b, c, d, e, and f) show UV absorptions.
|
Additional Problems
Visualizing Chemistry
| 14.16 |
|
| 14.17 |
|
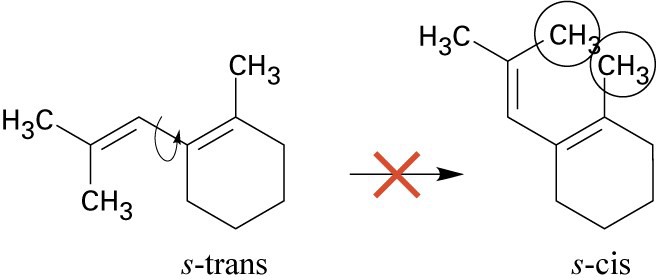
| 14.18 | In order to undergo Diels–Alder reaction, this s-trans diene would have to rotate to an s– cis arrangement. In an s-cis conformation, however, the two circled methyl groups experience steric strain by being too close to each other, preventing the molecule from adopting this conformation. Thus, Diels–Alder reaction doesn’t occur.
|
| 14.19 |
|
Mechanism Problems
| 14.20 | Note: The major product of the reaction depends on the reaction temperature. At higher temperatures (> 40 °C) the more substituted akene (the thermodynamic product) is favored, while at lower temperatures (0 °C) the less substituted alkene (the kinetic product) is favored.
|
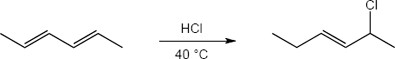
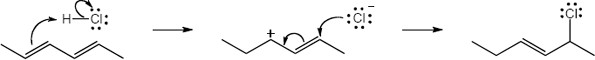
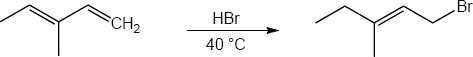
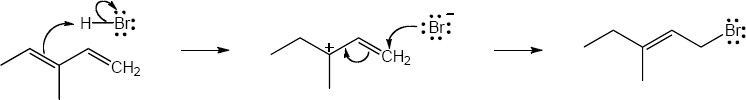
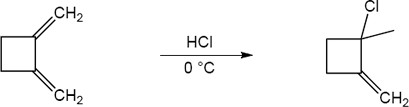
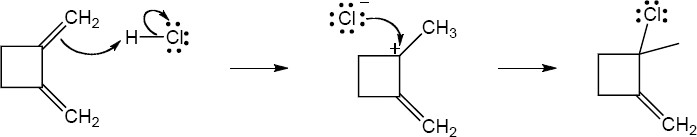
| 14.21 | Diels–Alder reactions are reversible when the products are much more stable (of lower energy) than the reactants. In this case, the reactant is a nonconjugated diene, and the products are benzene (a stable, conjugated molecule) and ethylene.
|
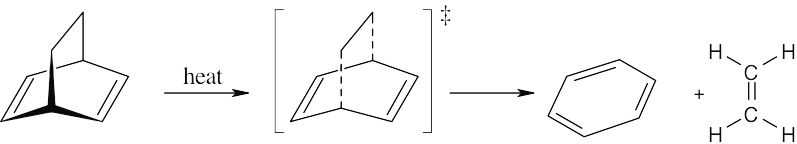
| 14.22 |
|
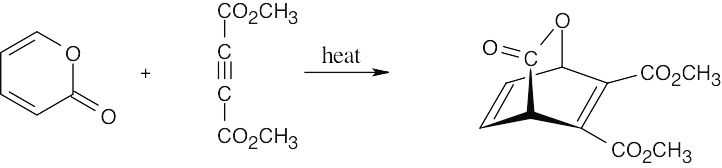
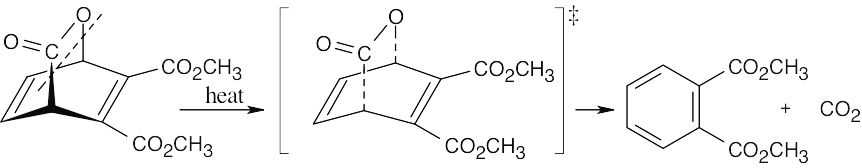
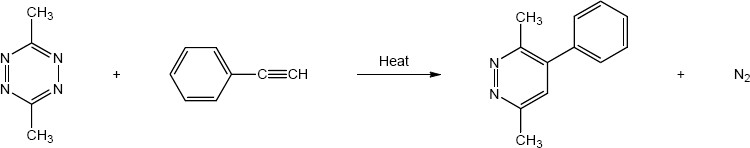
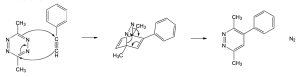
| 14.23 |
|
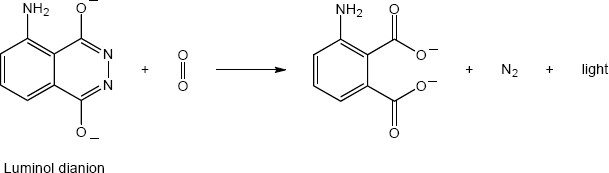
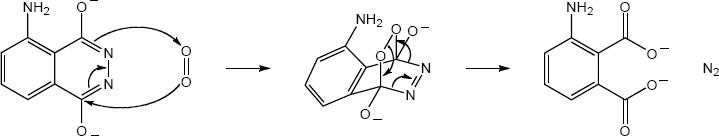
| 14.24 |
|
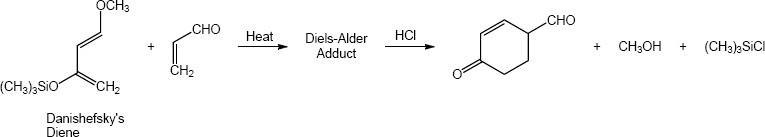
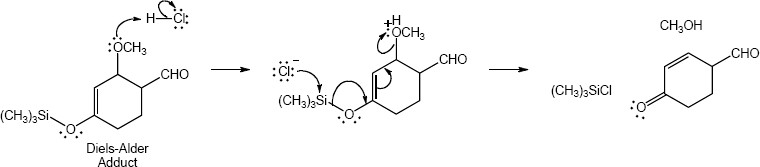
Conjugated Dienes
| 14.25 | All of these compounds can exhibit E/Z isomerism.
|
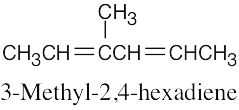
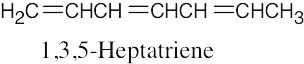
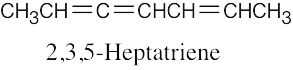
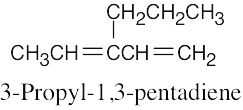
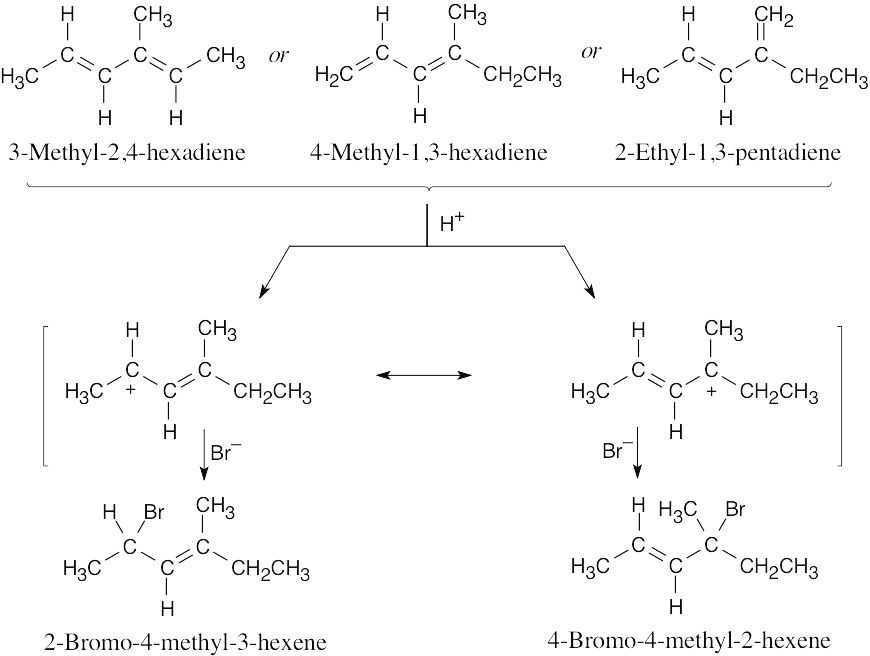
| 14.26 | Excluding double-bond isomers:
|
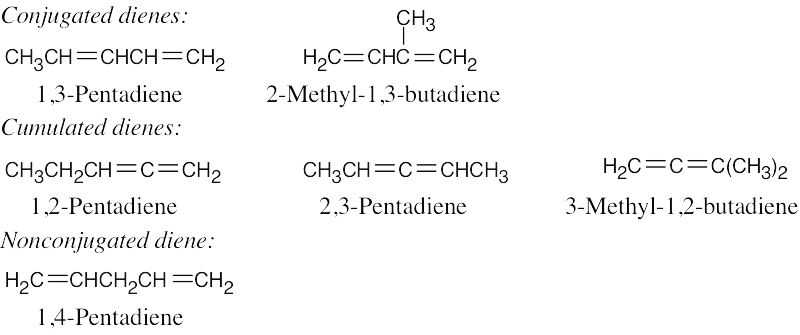
| 14.27 |
|
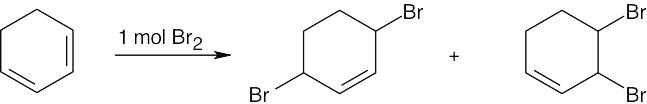
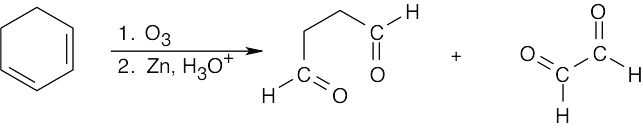
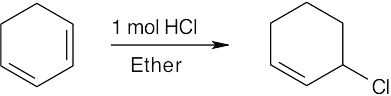
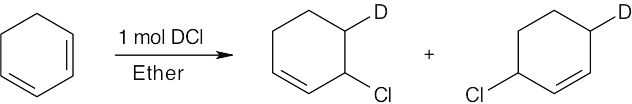
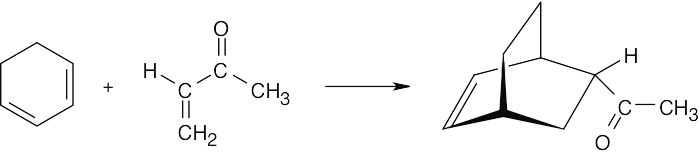
| 14.28 |
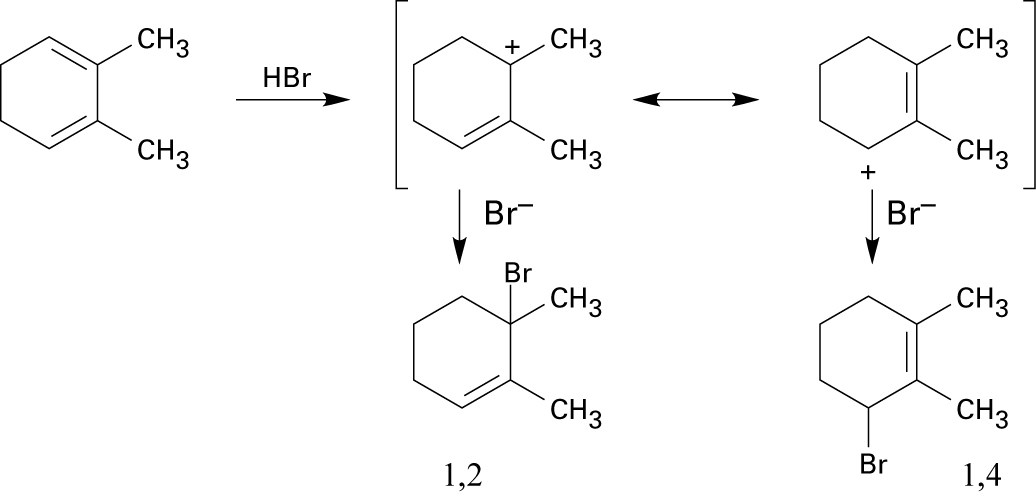
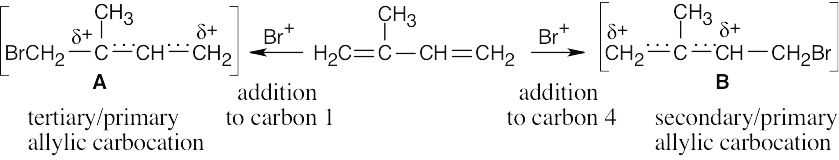
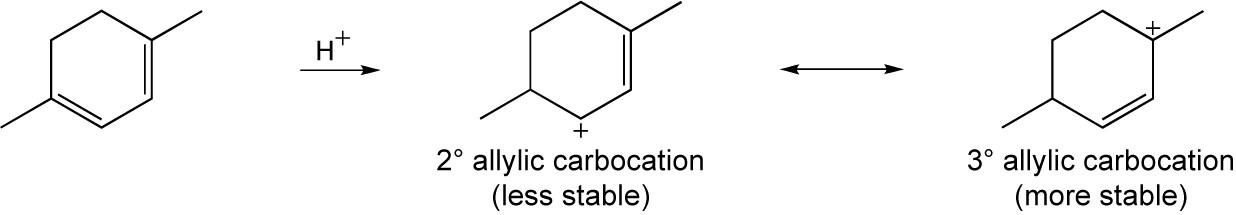
Tertiary/primary allylic carbocation A is more stable than secondary/primary allylic carbocation B. Since the products formed from the more stable intermediate predominate, 3,4-dibromo-3-methyl-1-butene is the major product of 1,2 addition of bromine to isoprene. In both cases, the product with the more substituted double bond (1,4 addition product) predominates. |
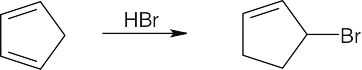
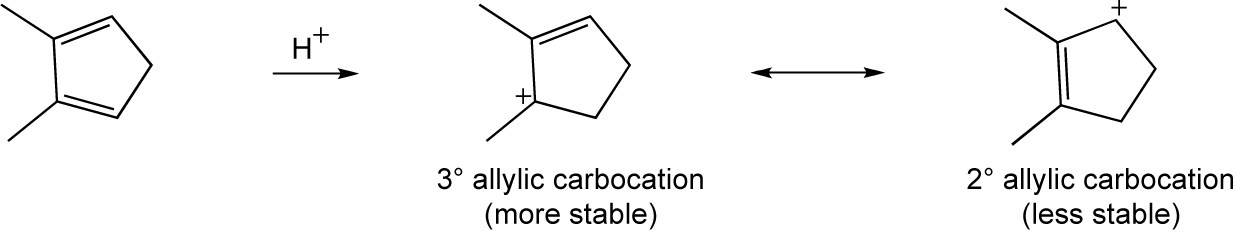
| 14.29 | Any unsubstituted cyclic 1,3-diene cyclic diene gives the same product from 1,2- and 1,4 addition. For example:
|
| 14.30 |
Carbocation D is most stable because it can use the π systems of both the benzene ring and the allylic side chain to delocalize positive charge. 3-Chloro-1-phenyl-1-butene is the major product because it results from cation D and because its double bond can be conjugated with the benzene ring to provide extra stability. |
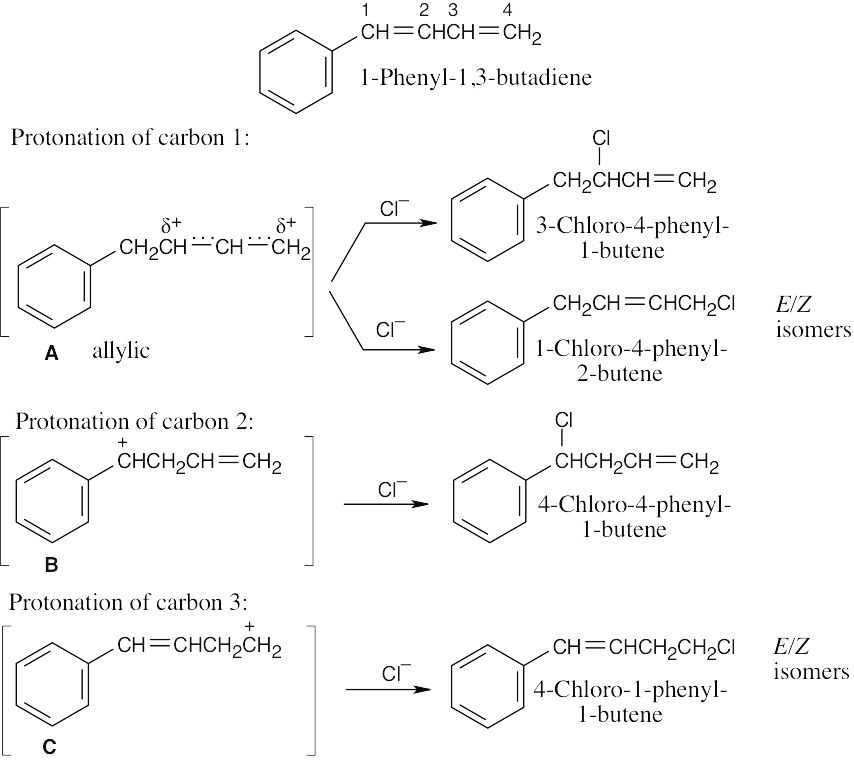
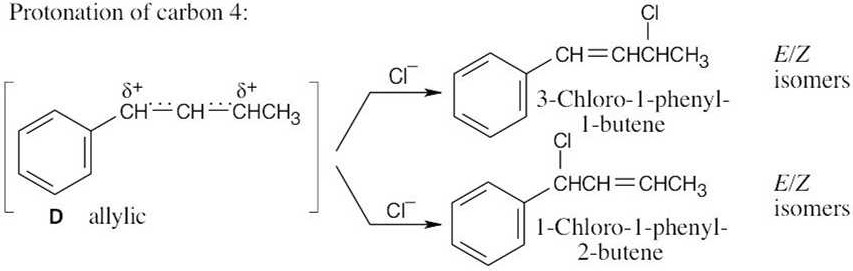
Diels–Alder Reactions
| 14.31 |
|
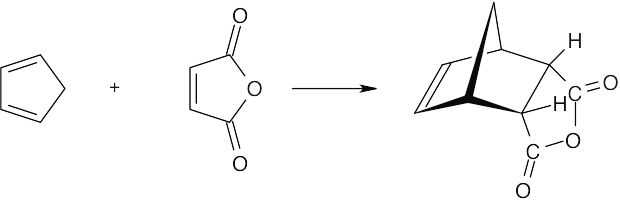
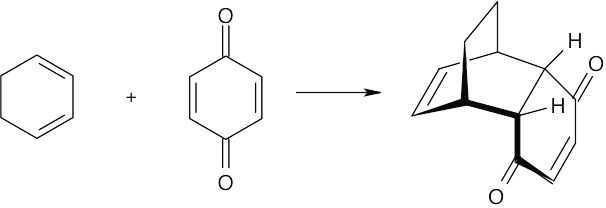
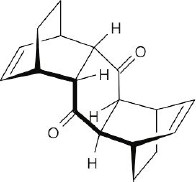
| 14.32 | This conformation of 2,3-di-tert-butyl-1,3-butadiene, in which the tert-butyl groups have an s-cis relationship, suffers from steric strain due to the bulky substituents. Instead, the molecule adopts the s-trans conformation, which relieves the strain but does not allow Diels–Alder reaction to take place. |
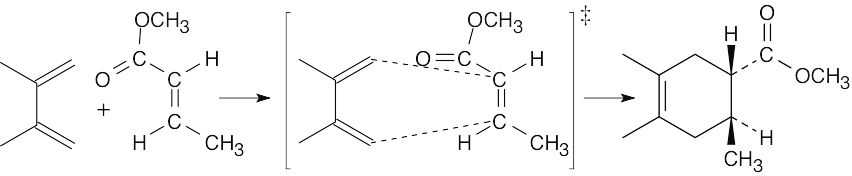
| 14.33 | The diene rotates to the s-cis conformation. The trans relationship of the two ester groups in the dienophile is preserved in the product.
|
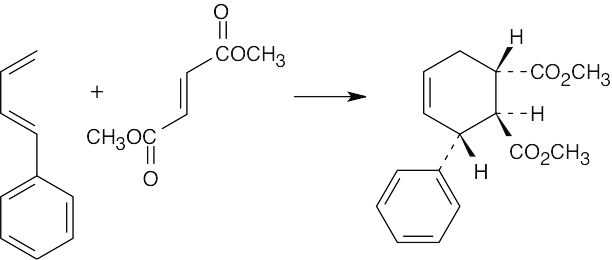
| 14.34 |
|
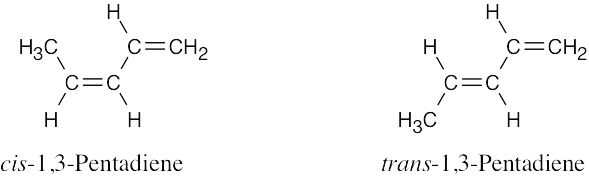

| 14.35 | HC≡CC≡CH can’t be used as a Diels–Alder diene because it is linear. The end carbons are too far apart to be able to react with a dienophile in a cyclic transition state.
Furthermore, the product of Diels–Alder addition would be impossibly strained, with two sp-hybridized carbons in a six-membered ring. |
| 14.36 |
Two different orientations of the dienophile ester group are possible, and two different products can form. |
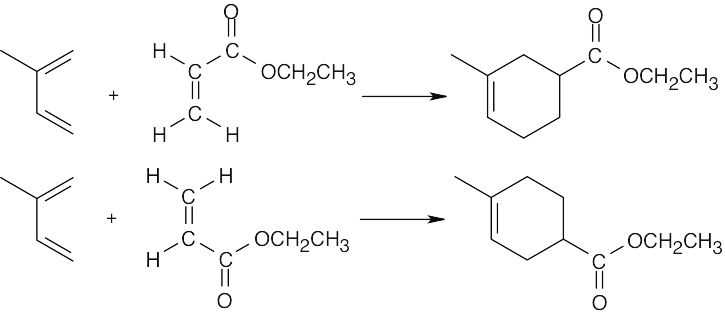
| 14.37 | The most reactive dienophiles contain electron-withdrawing groups.
Most reactive ————————> Least reactive (NC)2C=C(CN)2 > H2C=CHCHO > H2C=CHCH3 > (CH3)2C=C(CH3)2 Four electron- One electron- One electron- Four electron- withdrawing groups withdrawing group donating group donating groups The methyl groups of 2,3-dimethyl-2-butene also decrease reactivity for steric reasons. |
| 14.38 | The difference in reactivity of the three cyclic dienes is due to steric factors. As the non-diene part of the molecule becomes larger, the carbon atoms at the end of the diene portion of the ring are forced farther apart. Overlap with the π system of the dienophile in the cyclic transition state is poorer, and reaction is slower. |
| 14.39 | Although an electron-withdrawing group increases the reactivity of a dienophile, it decreases the reactivity of a diene. |
| 14.40 | First, find the cyclohexene ring formed by the Diels–Alder reaction. After you locate the new bonds, you should then be able to identify the diene and the dienophile.
|
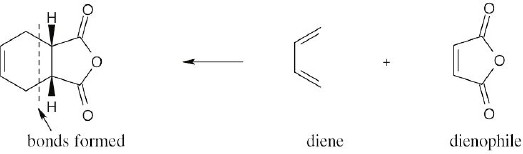
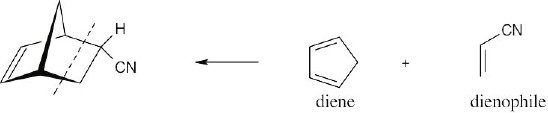
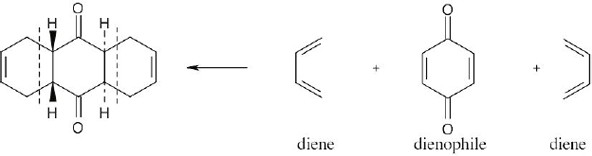
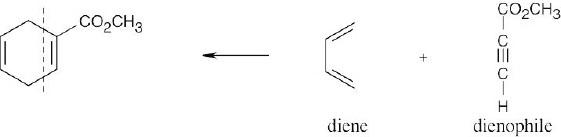
| 14.41 |
|
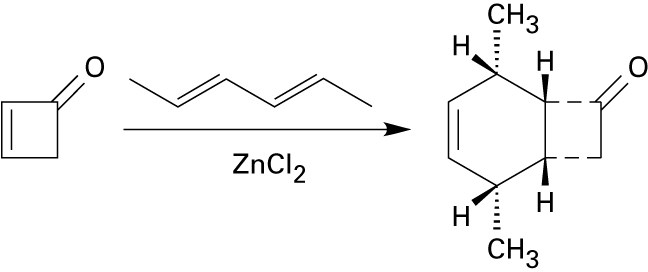
Diene Polymers
| 14.42 |
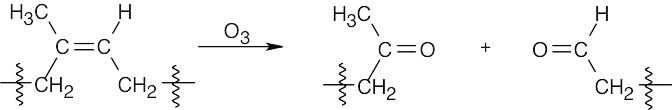
Ozone causes oxidative cleavage of the double bonds in rubber and breaks the polymer chain. |
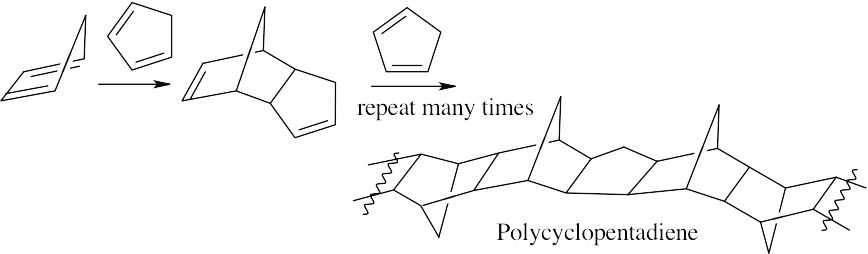
| 14.43 | Polycyclopentadiene is the product of successive Diels–Alder additions of cyclopentadiene to a growing polymer chain. Strong heat causes depolymerization of the chain and reformation of cyclopentadiene monomer units.
|
UV Spectroscopy
| 14.44 | Note: In general, the more conjugated double bonds that a molecule has, the longer the wavelength at which it will absorb UV light.
|
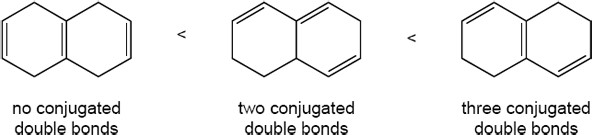
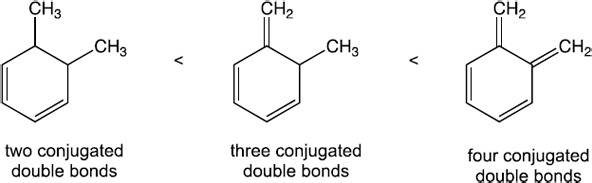
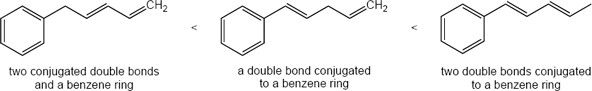
| 14.45 | Only compounds having alternating multiple bonds show π → π* ultraviolet absorptions in the 200–400 nm range. Of the compounds shown, only pyridine (b) absorbs in this range. |
| 14.46 | To absorb in the 200–400 nm range, an alkene must be conjugated. Since the double bonds of allene aren’t conjugated, allene doesn’t absorb light in the UV region. |
| 14.47 | The value of λmax in the ultraviolet spectrum of dienes becomes larger with increasing alkyl substitution. Since energy is inversely related to λmax, the energy needed to produce ultraviolet absorption decreases with increasing substitution.
Each alkyl substituent causes an increase in λmax of 3–6 nm. |
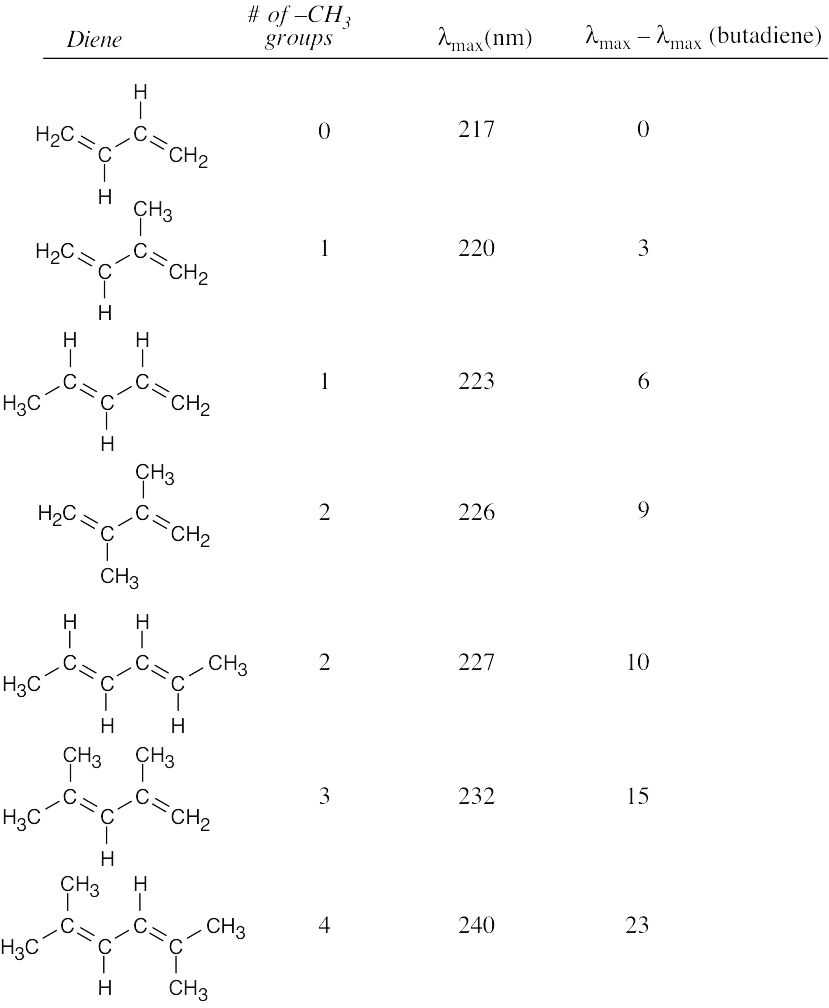
| 14.48 |
In Problem 14.41, we concluded that one alkyl group increases λmax of a conjugated diene by approximately 5 nm. Since 2,3-dimethyl-1,3,5-hexatriene has two methyl substituents, its UV λmax should be about 10 nm longer than the λmax of 1,3,5-hexatriene. |
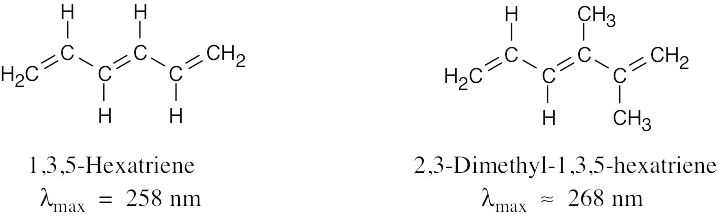
| 14.49 |
|
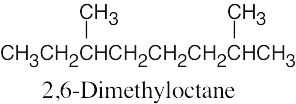

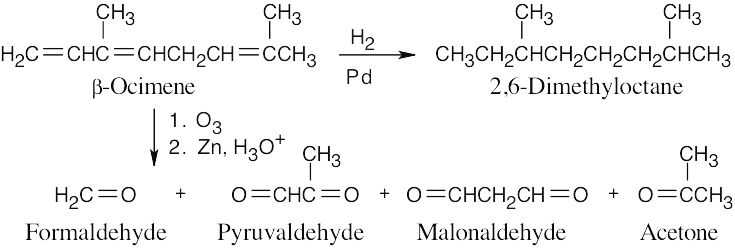
General Problems
| 14.50 |
|

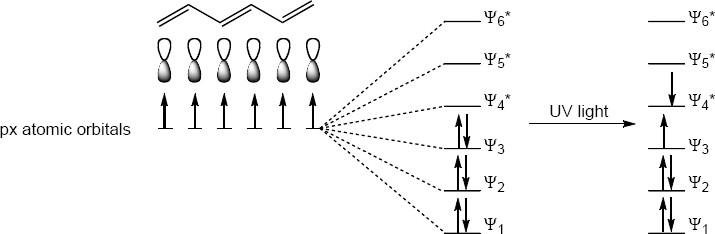
| 14.51 | All of the questions can be answered with the figure below.
(a) The three pi-bonds that are conjugated each have two p-atomic orbitals. Thus, there are six p-atomic orbitals in the conjugated system. (b) The total number of molecular orbitals must equal the number of atomic orbitals used to create them. Therefore there are six molecular orbitals. (Ψ1, Ψ2, Ψ3, Ψ4, Ψ5, Ψ6) (c) The bonding molecular orbitals are lower in energy than the p-atomic orbital energies. Thus, there are three bonding orbitals (Ψ1, Ψ2, Ψ3). (d) The antibonding molecular orbitals are higher in energy than the p-atomic orbital energies. Thus, there are three antibonding orbitals (Ψ4*, Ψ5*, Ψ6*; antibonding orbitals are denoted with an *). (e) Because orbitals fill from lowest energy to highest energy and there are six electrons in the bonding Ψ1, Ψ2 and Ψ3 molecular orbitals. (f) The electron would move from the Ψ3 (the highest occupied molecular orbital; HOMO) to Ψ4* (the lowest unoccupied molecular orbital; LUMO). |
| 14.52 |
2,4-Hexadiene can easily be distinguished from the other two isomers because it is the only isomer that absorbs in the UV region. The other two isomers show significant differences in their 1H and 13C NMR spectra and can be identified by either technique. |
| 14.53 |
There are two reasons why the other regioisomer is not formed: (1) Carbon 1 is less nucleophilic than carbon 2; (2) The cation intermediate that would result from protonation at carbon 1 can’t be stabilized by the oxygen electrons and does not form. |
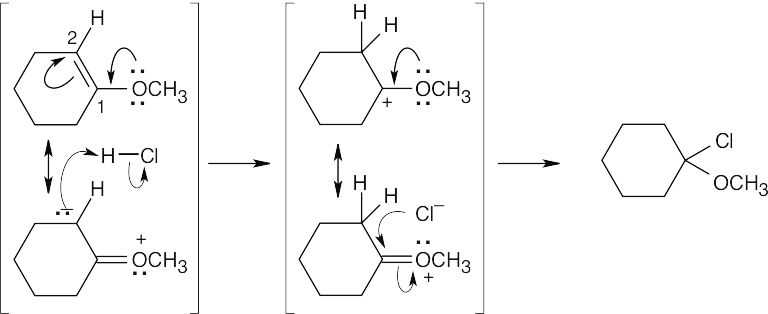
| 14.54 |
|
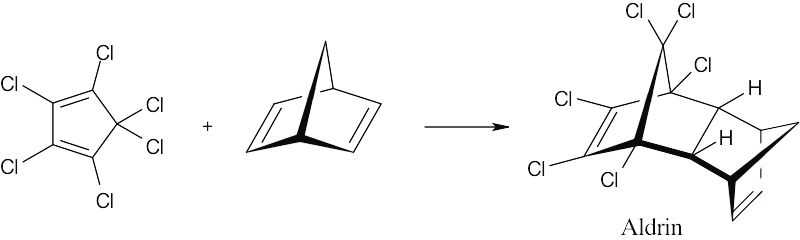
| 14.55 |
|
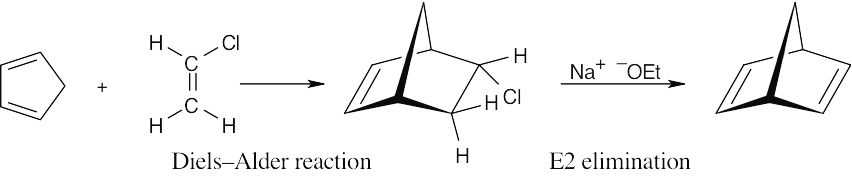
| 14.56 | The first equivalent of maleic anhydride adds to the s-cis bond of the triene.
|
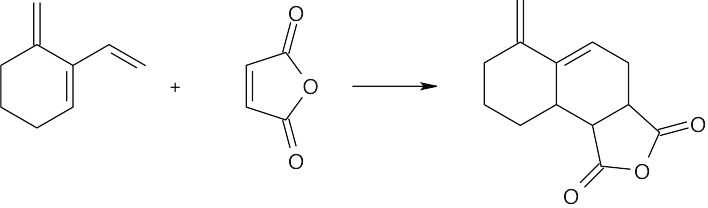
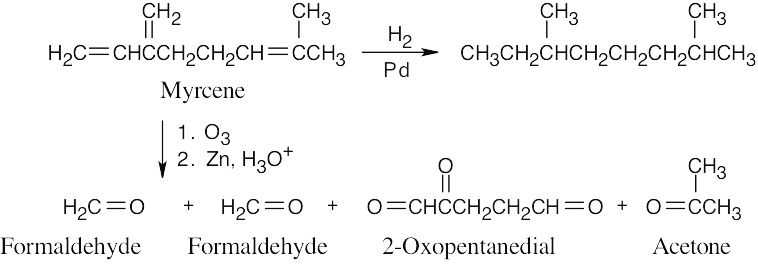
| 14.57 | Much of what was proven for β-ocimene (Problem 14.49) is also true for myrcene, since both hydrocarbons have the same carbon skeleton and contain conjugated double bonds. The difference between the two isomers is in the placement of the double bonds.
The ozonolysis fragments from myrcene are 2-oxopentanedial (five carbon atoms), acetone (three carbon atoms), and two equivalents of formaldehyde (one carbon atom each). Putting these fragments together in a manner consistent with the data gives the following structural formula for myrcene:
|
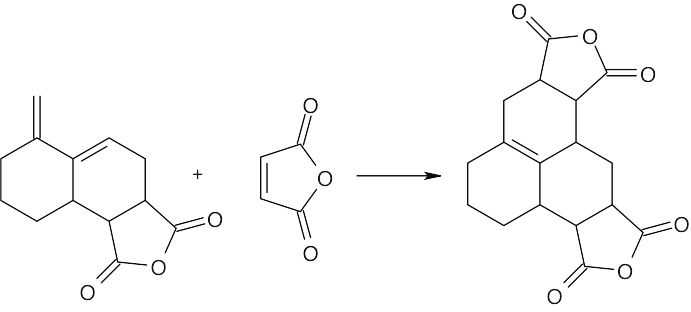
| 14.58 |
|
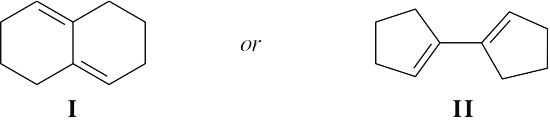
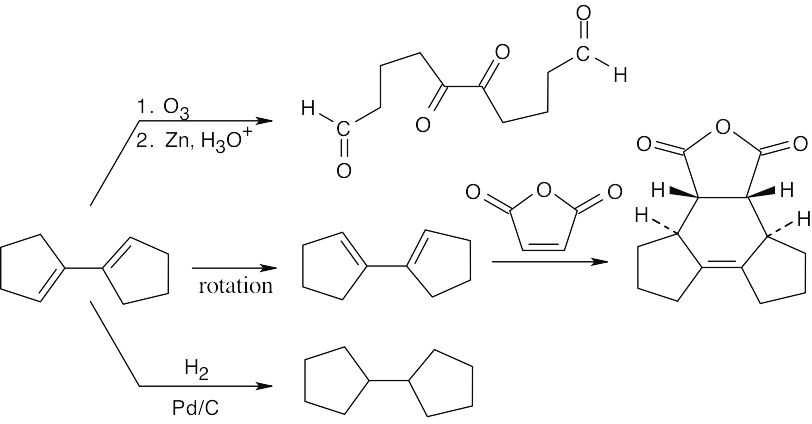
| 14.59 |
|
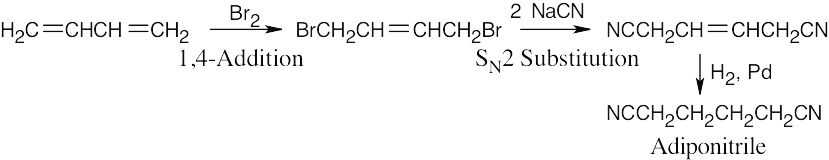
| 14.60 |
|

| 14.61 |
|
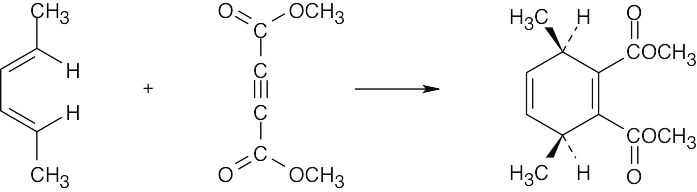
| 14.62 |
The stereochemistry of the product resulting from Diels–Alder reaction of the (2E,4Z) diene differs at the starred carbon from that of the (2E,4E) diene. Not only is the stereochemistry of the dienophile maintained during the Diels–Alder reaction, the stereochemistry of the diene is also maintained. |
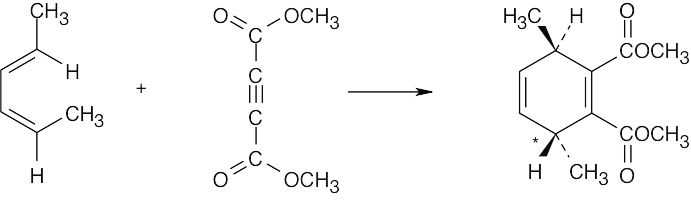
- Although it is usually best to work backwards in a synthesis problem, it sometimes helps to work both forwards and backwards. In this problem, we know that the starting materials are a diene and a dienophile. This suggests that the synthesis involves a Diels– Alder reaction. The product is a dialdehyde in which the two aldehyde groups have a cis relationship, indicating that they are the products of ozonolysis of a double bond that is part of a ring. These two pieces of information allow us to propose the following synthesis:
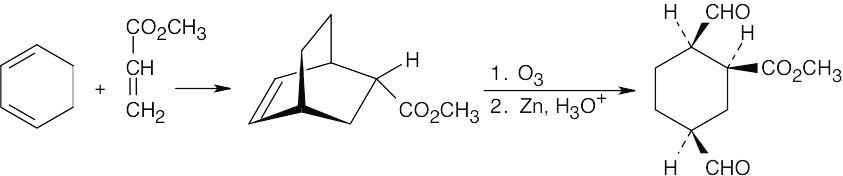
The –CHO groups are cis to the ester in the product.
- The lone pair electrons from nitrogen can overlap with the double bond π electrons in a manner similar to the overlap of the π electrons of two conjugated double bonds. This electron contribution from nitrogen makes an enamine double bond electron-rich.
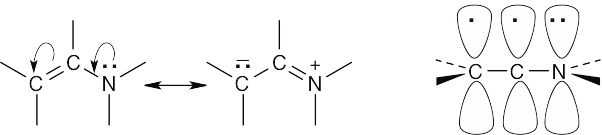
The orbital picture of an enamine shows a 4 p-electron system that resembles the system of a conjugated diene.
- Double bonds can be conjugated not only with other multiple bonds but also with the lone-pair electrons of atoms such as oxygen and nitrogen. p-Toluidine has the same number of double bonds as benzene, yet its λmax is 31 nm greater. The electron pair of the nitrogen atom can conjugate with the π electrons of the three double bonds of the ring, extending the π system and increasing λmax.
This file is copyright 2023, Rice University. All Rights Reserved.
