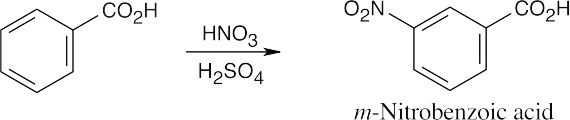
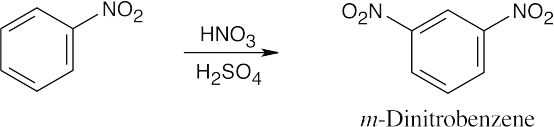
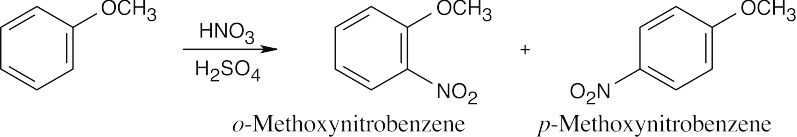
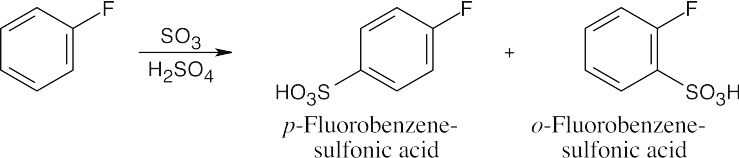
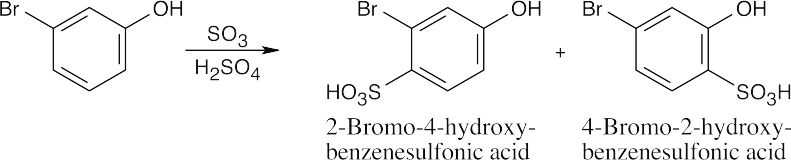
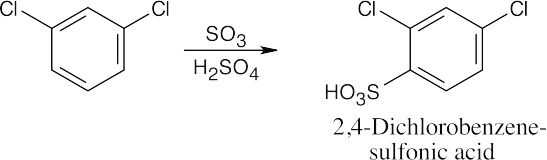
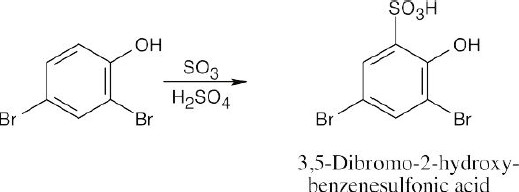
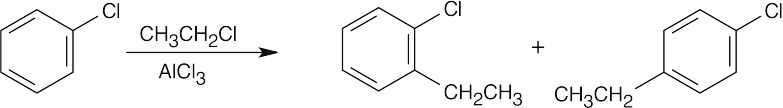
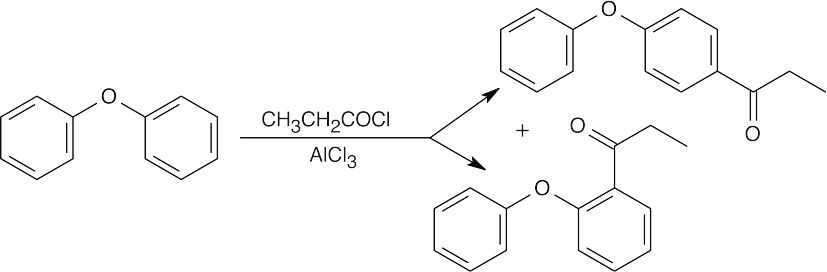
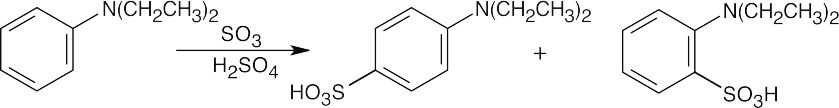
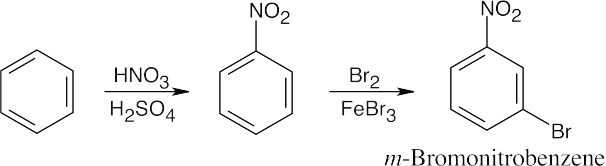
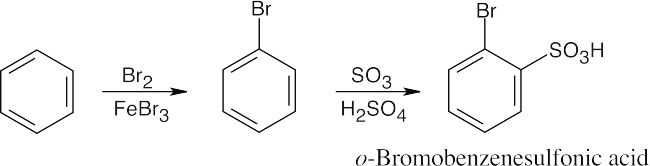
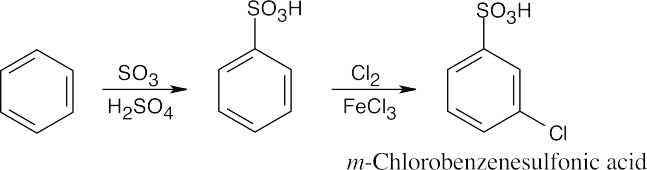
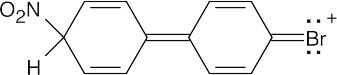
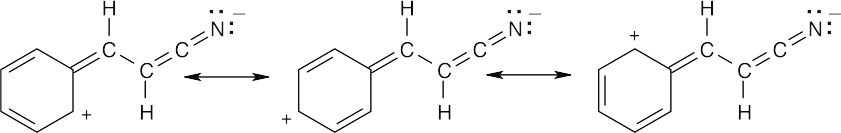
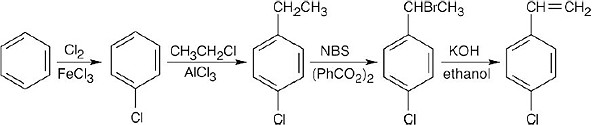
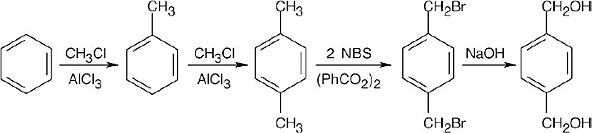
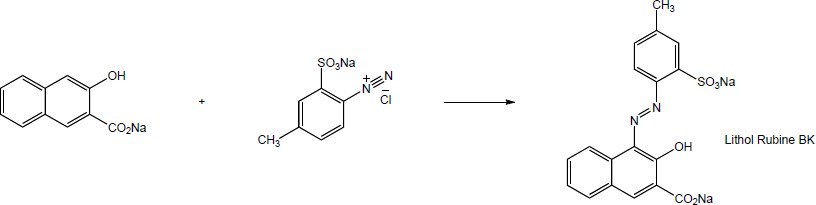
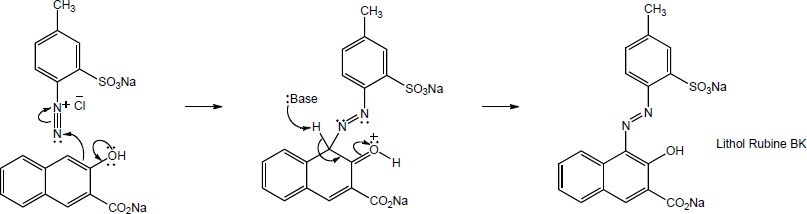
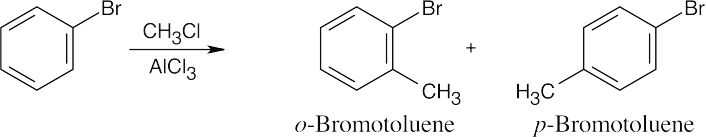
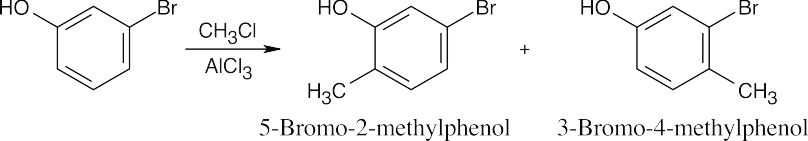
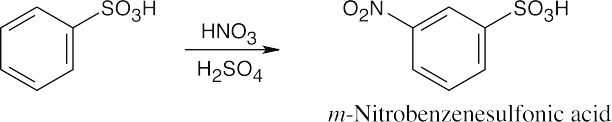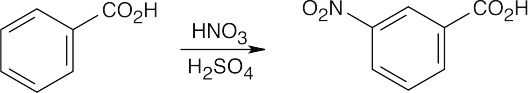16 Chapter 16 – Chemistry of Benzene: Electrophilic Aromatic Substitution Solutions to Problems
Chapter 16 – Chemistry of Benzene: Electrophilic Aromatic Substitution
Solutions to Problems
| 16.2 |
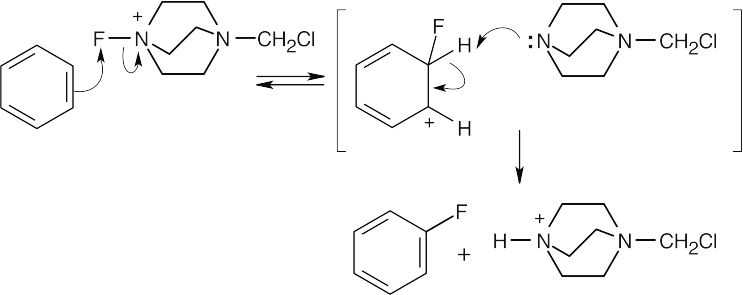
The π electrons of benzene attack the fluorine of F-TEDA-BF4, and the nonaromatic intermediate loses –H to give the fluorinated product. |
| 16.3 |
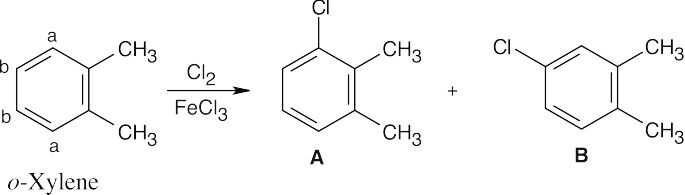
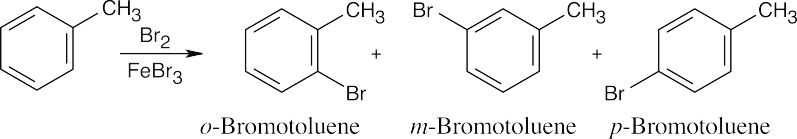
Chlorination at either position “a” of o-xylene yields product A, and chlorination at either position “b” yields product B.
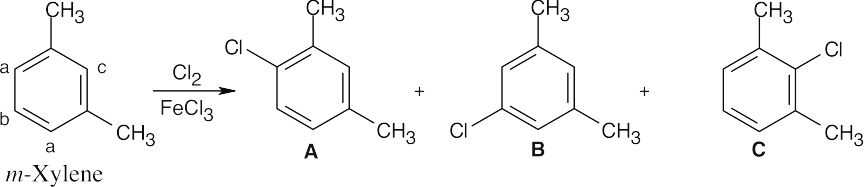
Three products might be expected to form on chlorination of m–xylene.
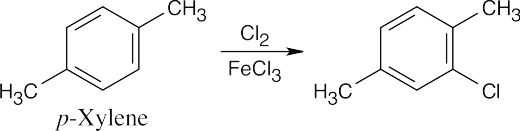
Only one product results from chlorination of p–xylene because all sites are equivalent. |
|
| 16.4 |
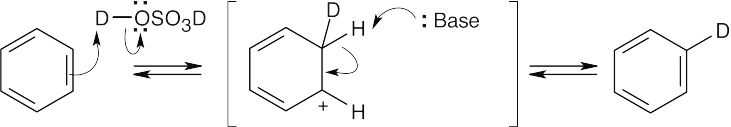
Benzene can be protonated by strong acids. The resulting intermediate can lose either deuterium or hydrogen. If –H is lost, deuterated benzene is produced. Attack by deuterium can occur at all positions of the ring and leads to eventual replacement of all hydrogens by deuterium. Only the first step is shown. |
| 16.5 |
Carbocation rearrangements of alkyl halides occur (1) if the initial carbocation is primary or secondary, and (2) if it is possible for the initial carbocation to rearrange to a more stable secondary or tertiary cation.
| (a) |
Although +CH2CH3 is a primary carbocation, it can’t rearrange to a more stable cation. |
| (b) |
CH3CH2CH(Cl)CH3 forms a secondary carbocation that doesn’t rearrange. |
| (c) |
+CH2CH2CH3 rearranges to the more stable +CH(CH3)2. |
| (d) |
+CH2C(CH3)3 (primary) undergoes an alkyl shift to yield (CH3)2C+CH2CH3 (tertiary) |
| (e) |
The cyclohexyl carbocation doesn’t rearrange. |
In summary:
No rearrangement: (a) CH3CH2Cl, (b) CH3CH2CH(Cl)CH3, (e) chlorocyclohexane
Rearrangement: (c) CH3CH2CH2Cl, (d) (CH3)3CCH2Cl |
| 16.6 |
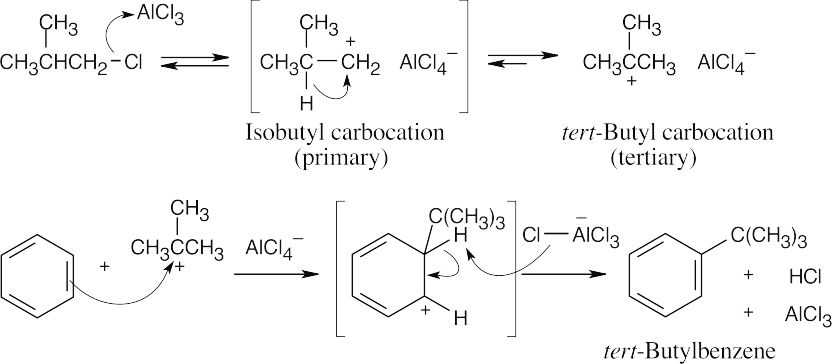
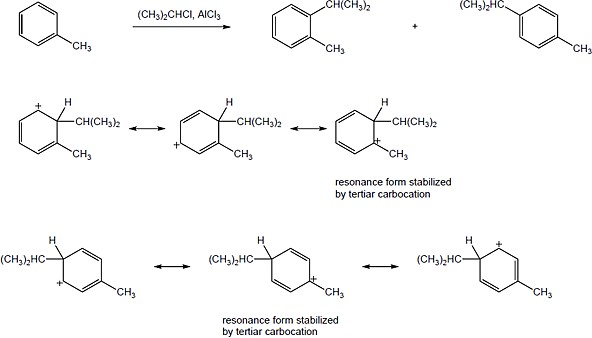
The isobutyl carbocation, initially formed when 1-chloro-2-methylpropane and AlCl3 react, rearranges via a hydride shift to give the more stable tert–butyl carbocation, which can then alkylate benzene to form tert–butylbenzene. |
| 16.7 |
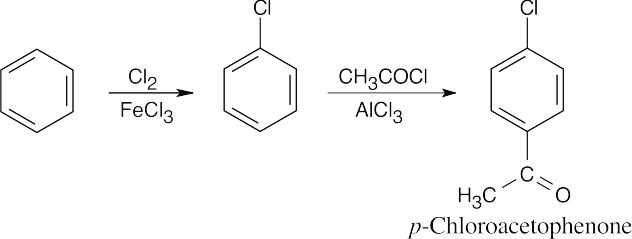
To identify the carboxylic acid chloride used in the Friedel–Crafts acylation of benzene, break the bond between benzene and the ketone carbon and replace it with a –Cl.
| (a)
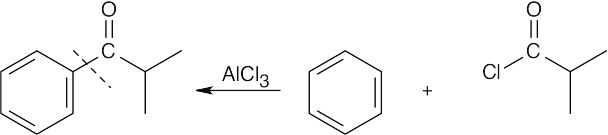
|
| (b)
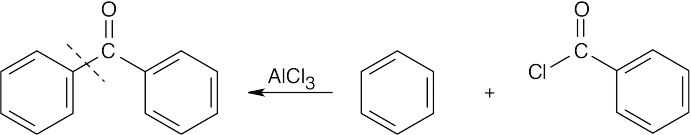
|
|
| 16.8 |
Use Figure 16.12 to find the activating and deactivating effects of groups.
Most Reactive ———————> Least Reactive
| (a) Phenol > toluene > benzene > nitrobenzene |
| (b) Phenol > benzene > chlorobenzene > benzoic acid |
| (c) Aniline > benzene > bromobenzene > benzaldehyde |
|
| 16.9 |
Refer to Figure 16.12 in the text for the directing effects of substituents. You should memorize the effects of the most important groups. As in Worked Example 16.2, identify the directing effect of the substituent, and draw the product.
| (a)
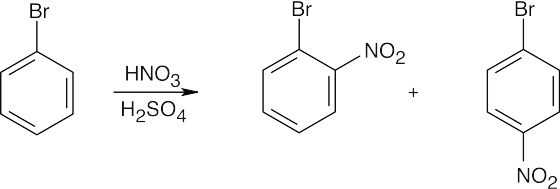
Even though bromine is a deactivator, it is an ortho-para director. |
| (b)
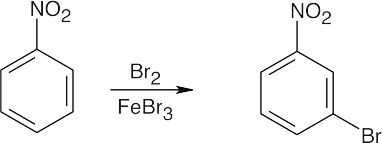
The –NO2 group is a meta-director. |
| (c)
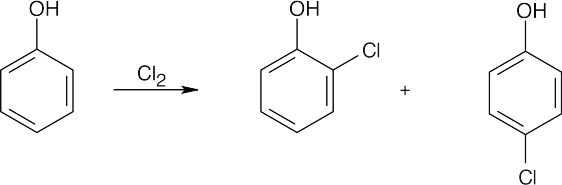
|
| (d)
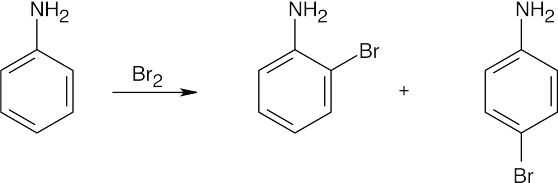
No catalyst is necessary because aniline is highly activating. |
|
| 16.10 |
An acyl substituent is deactivating. Once an aromatic ring has been acylated, it is much less reactive to further substitution. An alkyl substituent is activating, however, so an alkyl-substituted ring is more reactive than an unsubstituted ring, and polysubstitution occurs readily. |
| 16.11 |
(Trifluoromethyl) benzene is less reactive toward electrophilic substitution than toluene. The electronegativity of the three fluorine atoms causes the trifluoromethyl group to be electron-withdrawing and deactivating toward electrophilic substitution. The electrostatic potential map shows that the aromatic ring of (trifluoromethyl) benzene is more electron-poor, and thus less reactive, than the ring of toluene (red). |
| 16.12 |
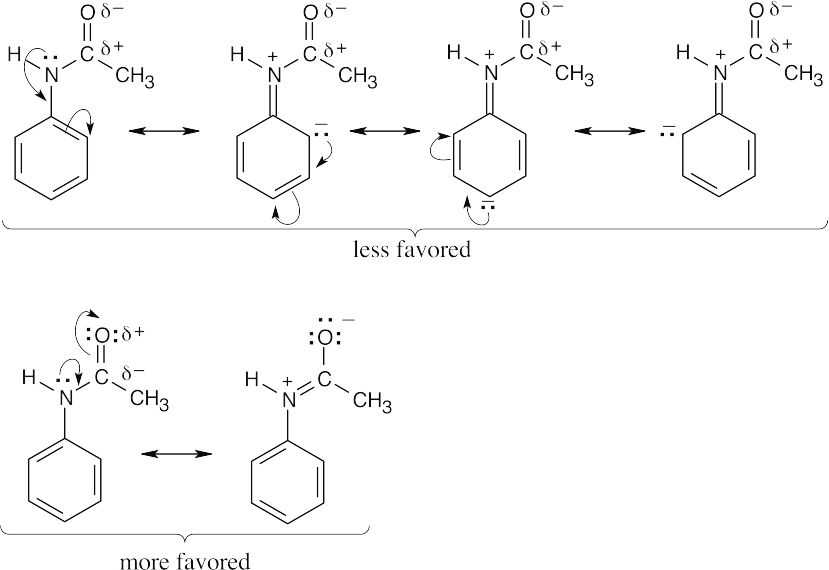
For acetanilide, resonance delocalization of the nitrogen lone pair electrons to the aromatic ring is less favored because the positive charge on nitrogen is next to the positively polarized carbonyl group. Resonance delocalization to the carbonyl oxygen is favored because of the electronegativity of oxygen. Since the nitrogen lone pair electrons are less available to the ring than in aniline, the reactivity of the ring toward electrophilic substitution is decreased, and acetanilide is less reactive than aniline toward electrophilic substitution. |
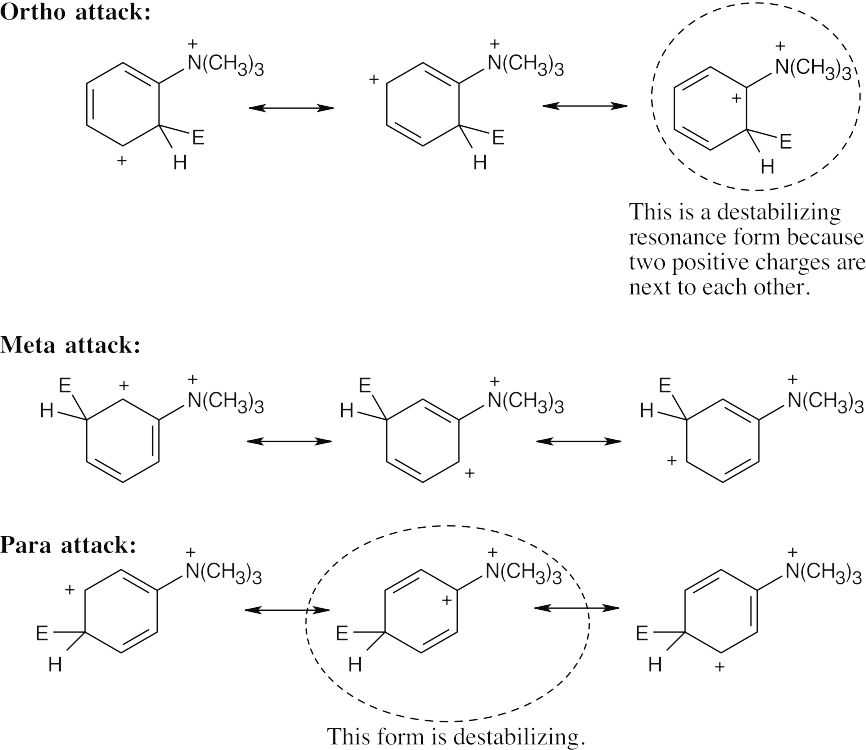
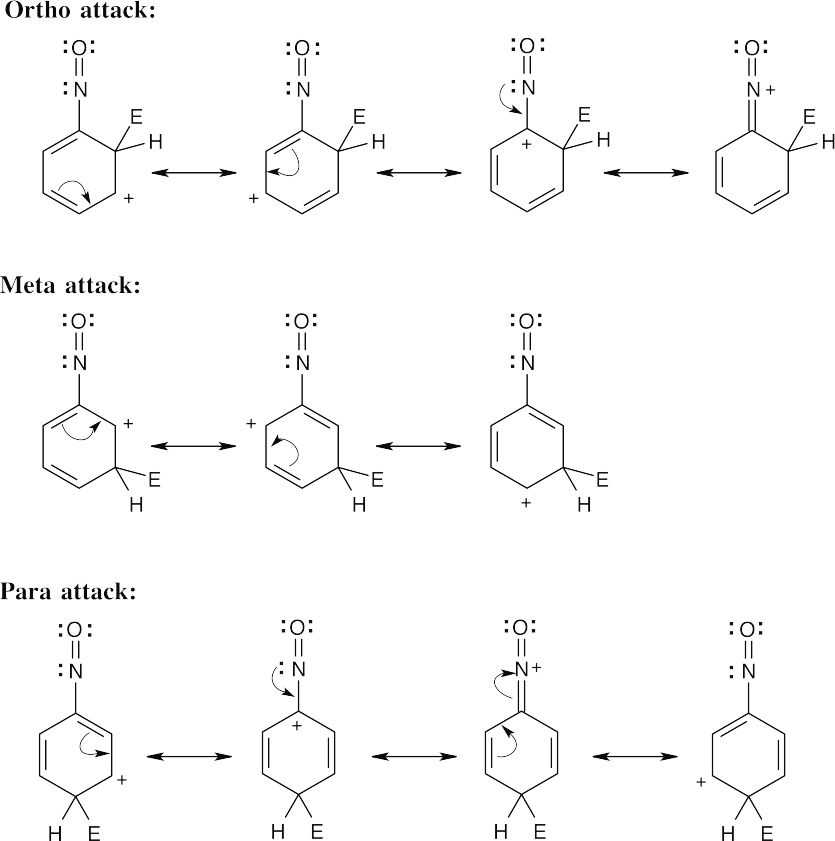
| 16.13 |
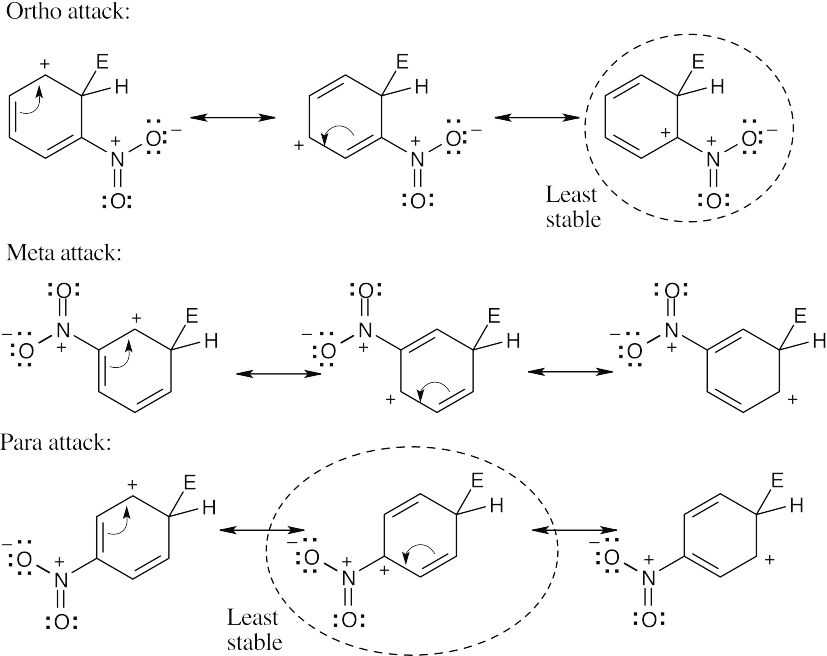
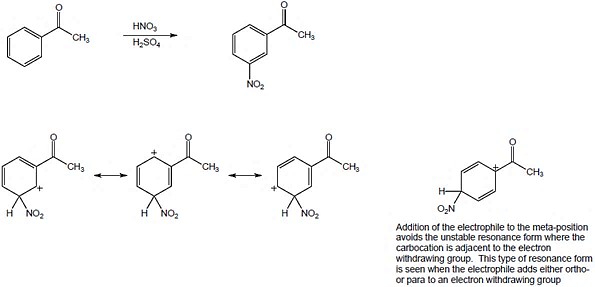
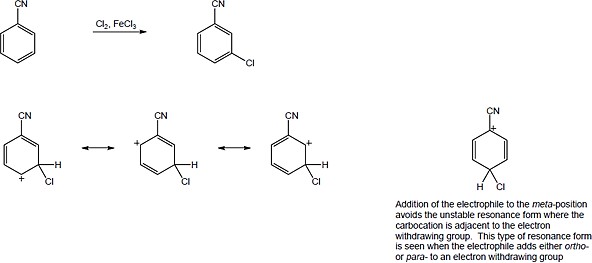
The circled resonance forms are unfavorable, because they place two positive charges adjacent to each other. The intermediate from meta attack is thus favored. |
| 16.14 |
| (a)
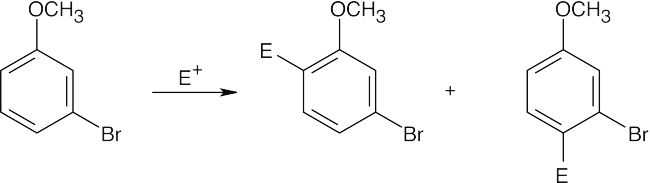
Both groups are ortho, para directors and direct substitution to the same positions.
Attack doesn’t occur between the two groups for steric reasons. |
| (b)
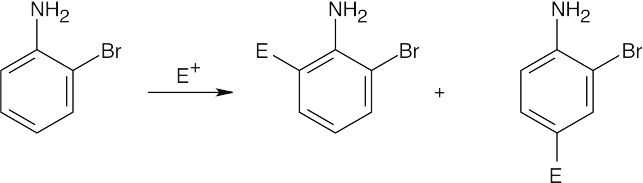
Both groups are ortho, para directors, but direct to different positions. Because –NH2 group is a more powerful activator, substitution occurs ortho and para to it. |
| (c)
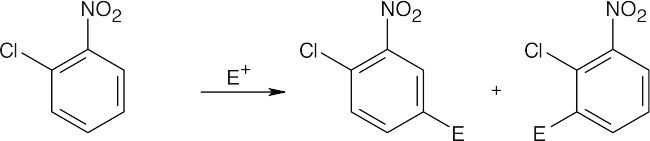
Both groups are deactivating, but they orient substitution toward the same positions. |
|
| 16.15 |
| (a)
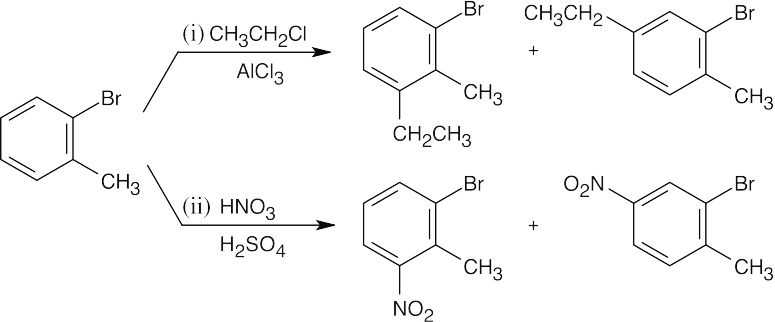
Although both groups are ortho, para directors, the methyl group directs the orientation of the substituents because it is a stronger activating group than bromine. |
| (b)
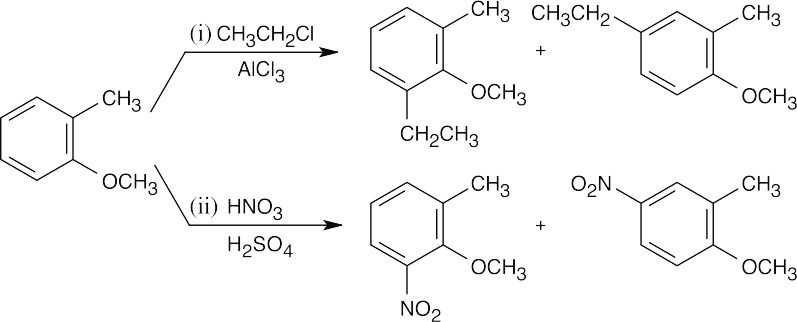
The methoxyl group directs substitution to the positions ortho and para to it. |
|
| 16.16 |
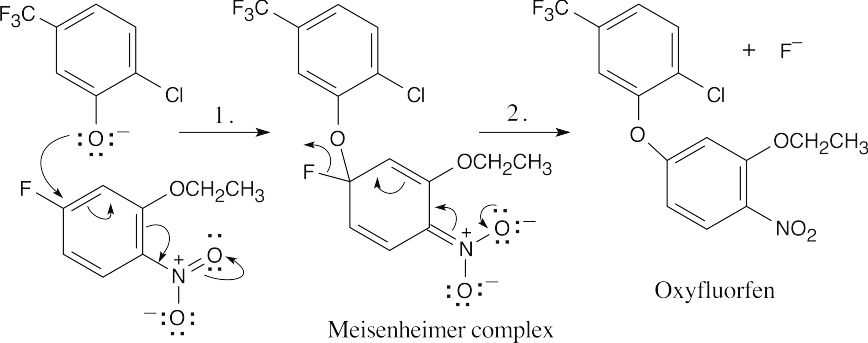
Hydroxide is used to form the nucleophilic phenoxide anion.
Step 1: Addition of the nucleophile.
Step 2: Elimination of fluoride ion.
The nitro group makes the ring electron-poor and vulnerable to attack by the nucleophilic RO– group. It also stabilizes the negatively charged Meisenheimer complex. |
| 16.17 |
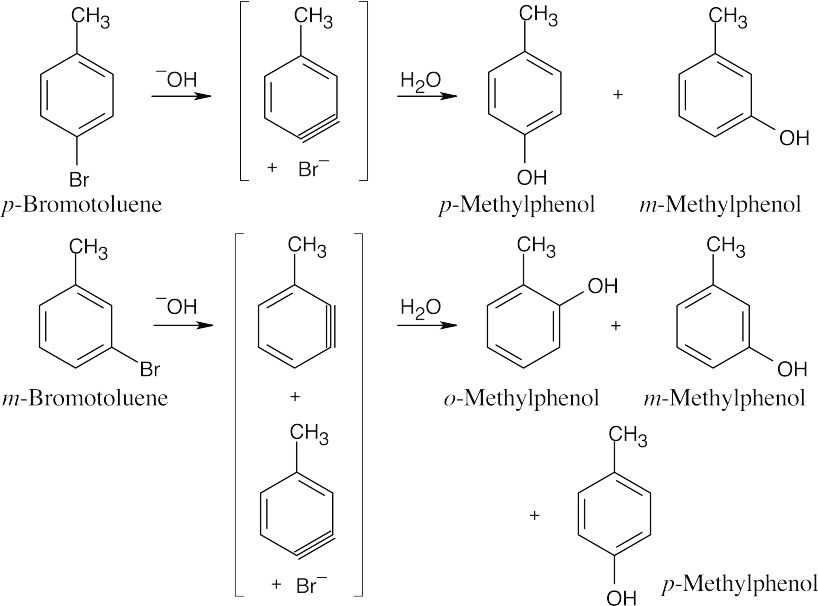
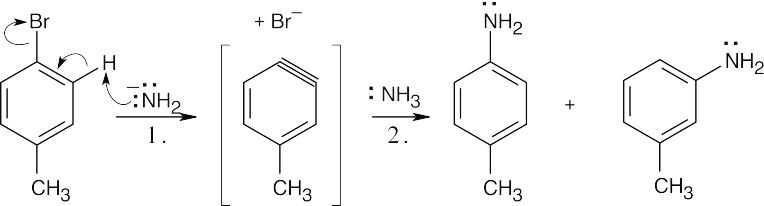
Treatment of m-bromotoluene with NaOH leads to two possible benzyne intermediates, which react with water to yield three methylphenol products. |
| 16.18 |
Oxidation takes place at the benzylic position.
| (a)
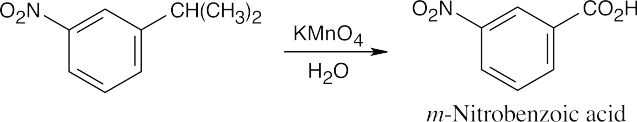
|
| (b)
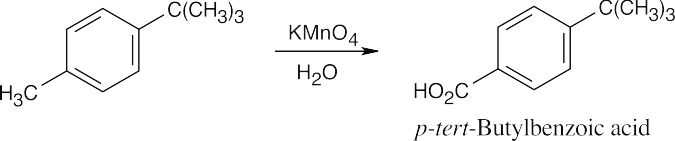
|
Treatment with KMnO4 oxidizes the methyl group but leaves the tert-butyl group untouched. |
| 16.19 |
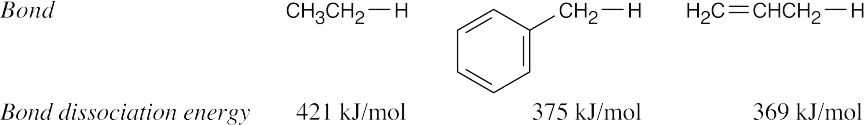
Bond dissociation energies measure the amount of energy that must be supplied to cleave a bond into two radical fragments. A radical is thus higher in energy and less stable than the compound from which it came. Since the C–H bond dissociation energy is 421 kJ/mol for ethane and 375 kJ/mol for a methyl group C–H bond of toluene, less energy is required to form a benzyl radical than to form an ethyl radical. A benzyl radical is thus more stable than a primary alkyl radical by 46 kJ/mol. The bond dissociation energy of an allyl C–H bond is 369 kJ/mol, indicating that a benzyl radical is nearly as stable as an allyl radical. |
| 16.22 |
| (a) |
In order to synthesize the product with the correct orientation of substituents, benzene must be nitrated before it is chlorinated.
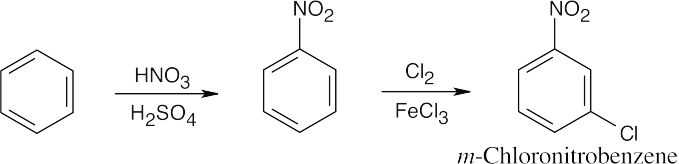
|
| (b) |
Chlorine can be introduced into the correct position if benzene is first acylated. The chlorination product can then be reduced.
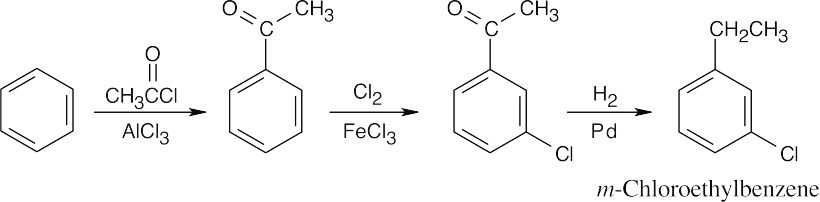
|
| (c) |
Friedel–Crafts acylation, followed by chlorination, reduction, and nitration, is the only route that gives a product in which the alkyl group and chlorine have a meta relationship.
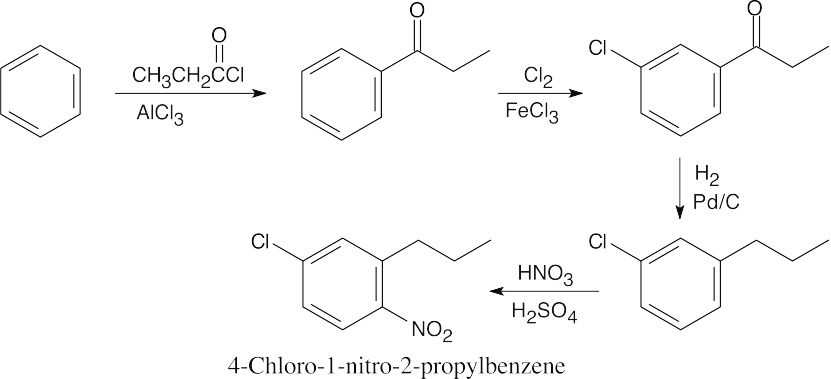
|
| (d) |
In planning this pathway, remember that the ring must be sulfonated after Friedel– Crafts alkylation because a sulfonated ring is too deactivated for alkylation to occur. Performing the reactions in this order allows the first two groups to direct bromine to the same position.
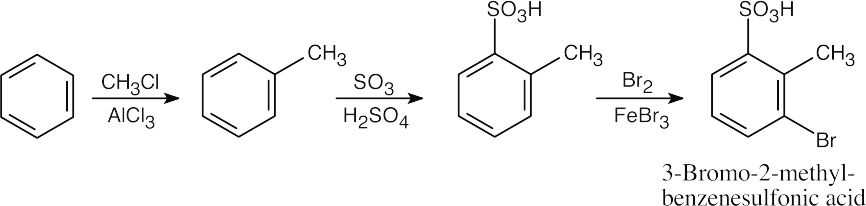
|
|
| 16.23 |
| (a) |
Friedel–Crafts acylation, like Friedel–Crafts alkylation, does not occur at an aromatic ring carrying a strongly electron-withdrawing group. |
| (b) |
There are two problems with this synthesis as it is written:
- Rearrangement often occurs during Friedel–Crafts alkylations using primary halides.
- Even if p-chloropropylbenzene could be synthesized, introduction of the second –Cl group would occur ortho, not meta, to the alkyl group.
A possible route to this compound:
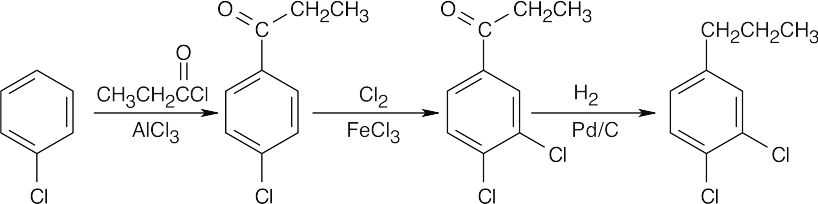
|
|
Additional Problems
Visualizing Chemistry
| 16.24 |
| (a) The methoxyl group is an ortho–para director.
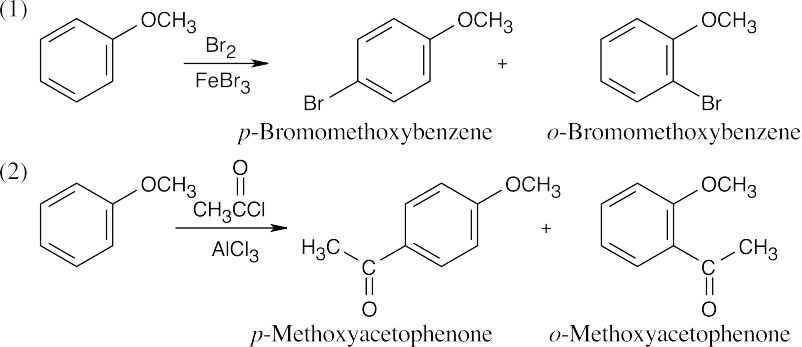
|
| (b) Both functional groups direct substituents to the same position.
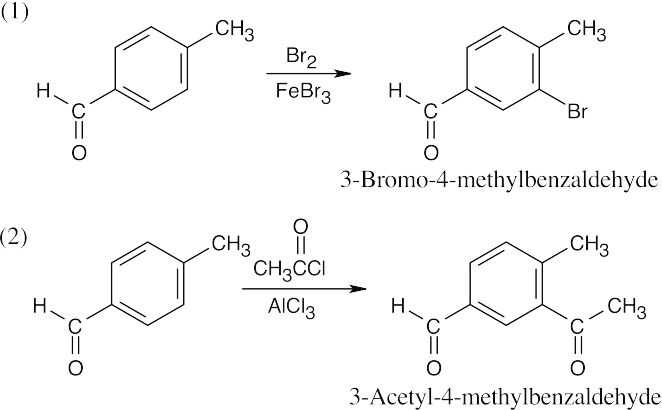
|
|
| 16.25 |
In the lowest-energy conformation of this biphenyl, the aromatic rings are tilted. If the rings had a planar relationship, steric strain between the methyl groups and the ring hydrogens on the second ring would occur. Complete rotation around the single bond doesn’t take place because the repulsive interaction between the methyl groups causes a barrier to rotation. |
| 16.27 |
Imagine two routes for synthesis of m-nitrotoluene:
| (1) Alkylation of benzene, followed by nitration, doesn’t succeed because an alkyl group is an o,p-director. |
| (2) Nitration of benzene, followed by alkylation, doesn’t succeed because nitrobenzene is unreactive to Friedel–Crafts alkylation. |
Thus, it isn’t possible to synthesize m-nitrotoluene by any route that we have studied in this chapter. |
Mechanism Problems
Mechanisms of Electrophilic Substitutions
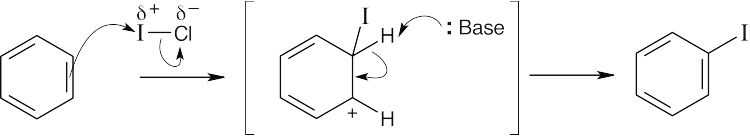
| 16.28 |
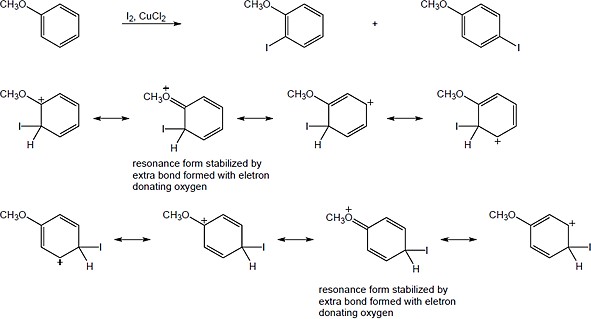
ICl can be represented as Iδ+ — Clδ− because chlorine is a more electronegative element than iodine. Iodine can act as an electrophile in electrophilic aromatic substitution reactions. |
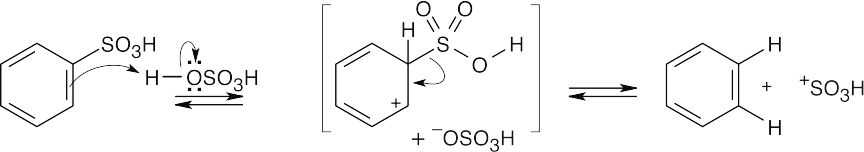
| 16.29 |
This mechanism is the reverse of the sulfonation mechanism illustrated in the text. H+ is the electrophile in this reaction. |
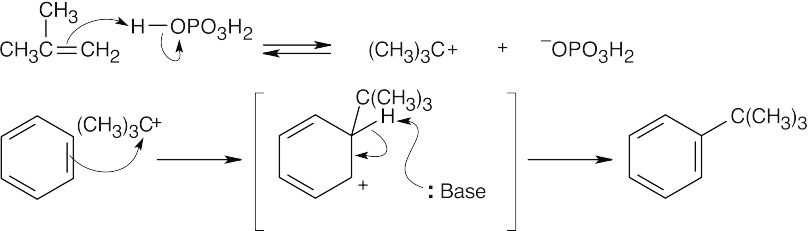
| 16.30 |
Phosphoric acid protonates 2-methylpropene, forming a tert-butyl carbocation. This carbocation acts as an electrophile in a Friedel–Crafts reaction to yield tert-butylbenzene. |
| 16.31 |
When an electrophile reacts with an aromatic ring bearing a (CH3)3N+– group:
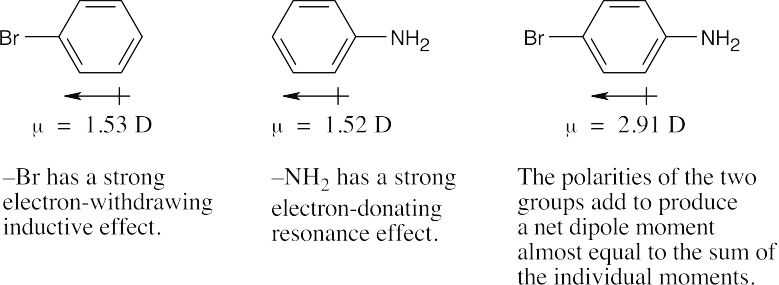
The N,N,N-trimethylammonium group has no electron-withdrawing resonance effect because it has no vacant p orbitals to overlap with the π orbital system of the aromatic ring. The (CH3)3N+– group is inductively deactivating, however, because it is positively charged. It is meta-directing because the cationic intermediate resulting from meta attack is somewhat more stable than those resulting from ortho or para attack. |
| 16.32 |
The aromatic ring is deactivated toward electrophilic aromatic substitution by the combined electron-withdrawing inductive effect of electronegative nitrogen and oxygen. The lone pair of electrons of nitrogen can, however, stabilize by resonance the ortho and para substituted intermediates but not the meta intermediate.
|
| 16.33 |
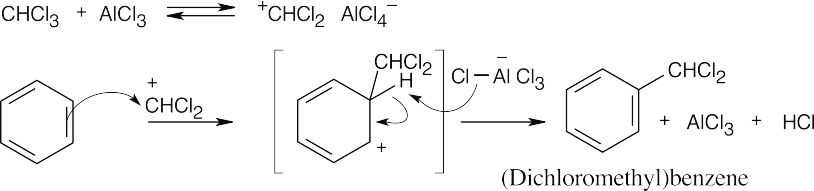
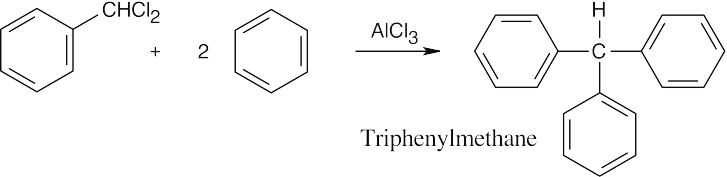
(Dichloromethyl) benzene can react with two additional equivalents of benzene by the same mechanism to produce triphenylmethane.
|
|
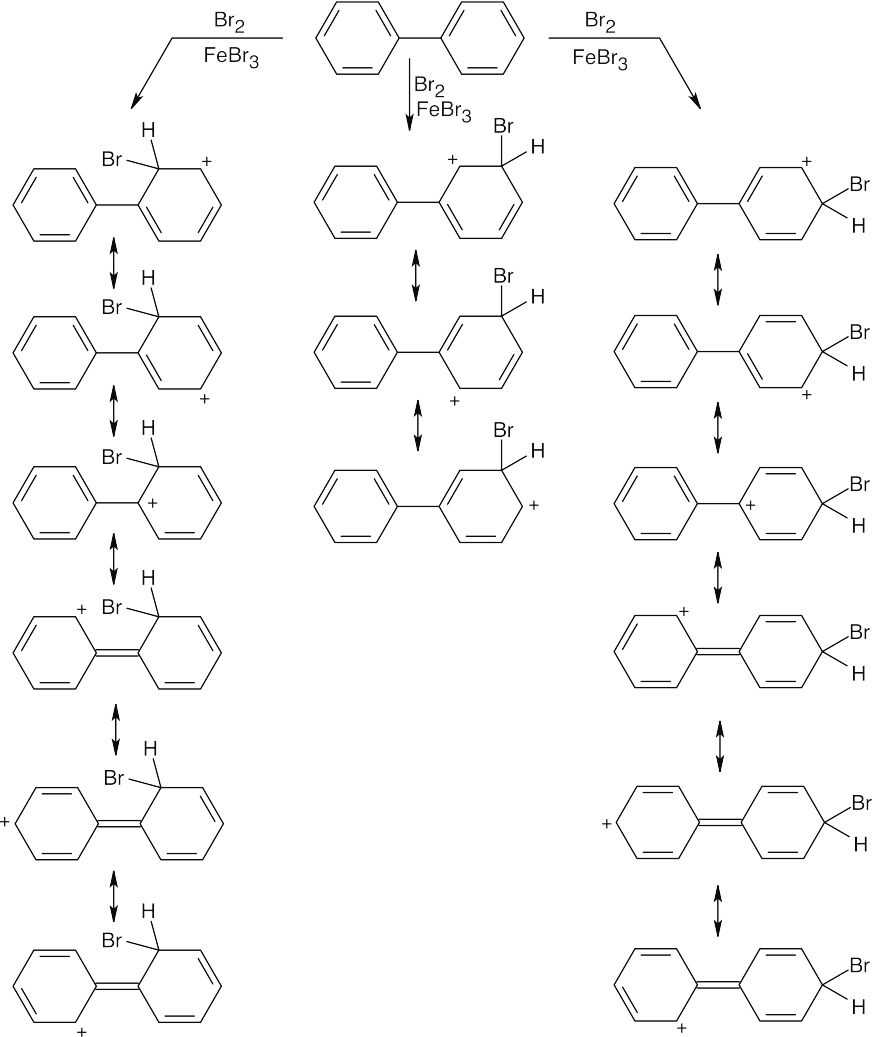
| 16.34 |
Resonance structures show that bromination occurs in the ortho and para positions of the rings. The positively charged intermediate formed from ortho or para attack can be stabilized by resonance contributions from the second ring of biphenyl, but this stabilization is not possible for meta attack. |
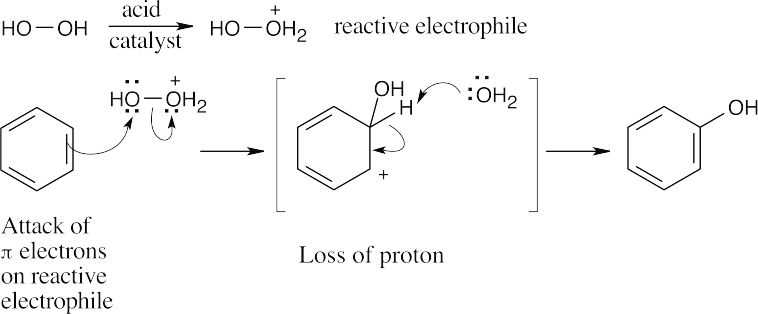
| 16.35 |
The reactive electrophile (protonated H2O2) is equivalent to +OH. |
Additional Mechanism Practice
| 16.36 |
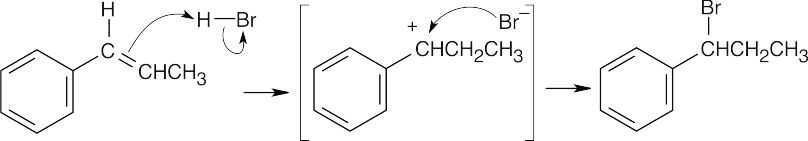
Protonation of the double bond at carbon 2 of 1-phenylpropene leads to an intermediate that can be stabilized by resonance involving the benzene ring. |
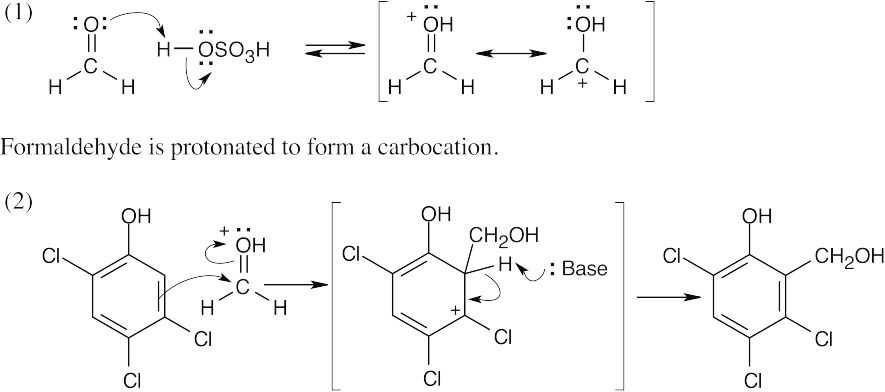
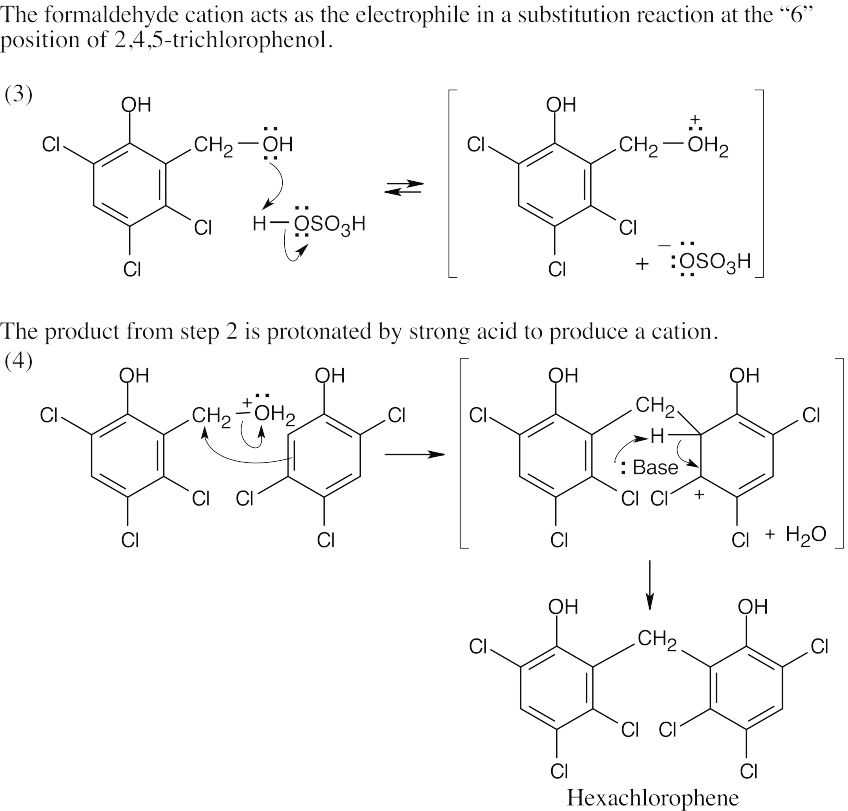
| 16.37 |
This cation is attacked by a second molecule of 2,4,5-trichlorophenol to produce hexachlorophene. |
| 16.39 |
Step 1: Attack of the nucleophile diethylamine.
Step 2: Loss of proton.
Step 3: Loss of Cl–.
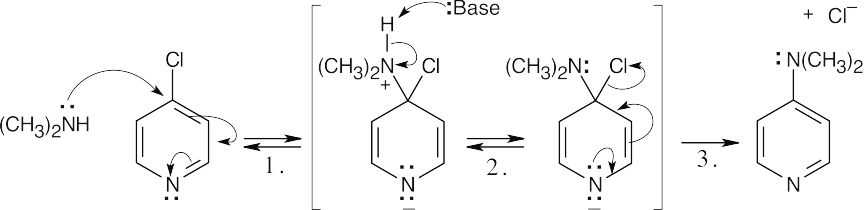
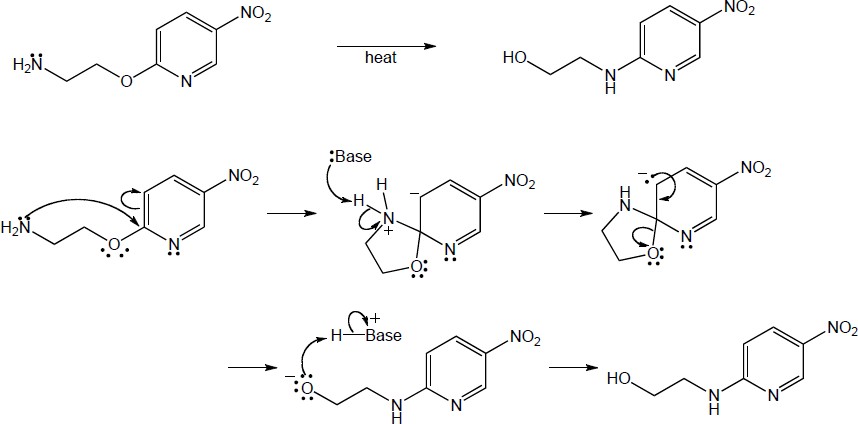
This reaction is an example of nucleophilic aromatic substitution. Dimethylamine is a nucleophile, and the pyridine nitrogen acts as an electron-withdrawing group that can stabilize the negatively-charged intermediate. |
| 16.40 |
Step 1: Formation of primary carbocation.
Step 2: Rearrangement to a secondary carbocation.
Step 3: Attack of ring π electrons on the carbocation.
Step 4: Loss of H+.
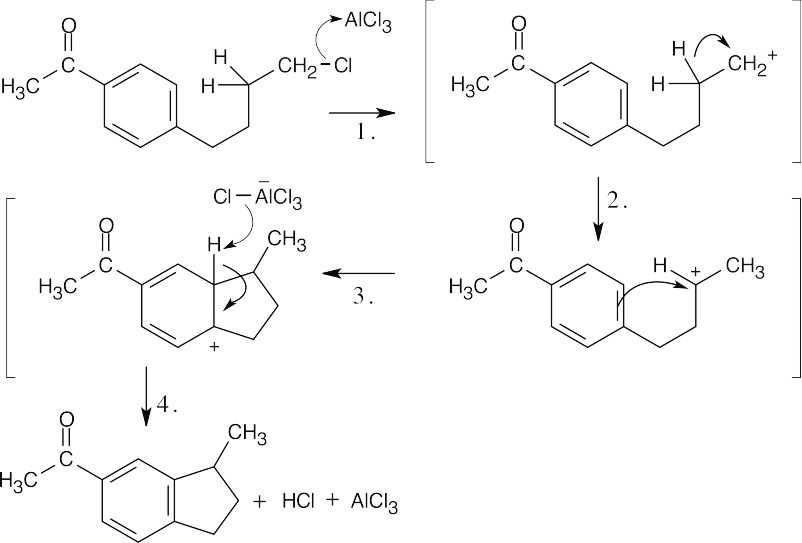
This reaction takes place despite the fact that an electron-withdrawing group is attached to the ring. Apparently, the cyclization reaction is strongly favored. |
| 16.41 |
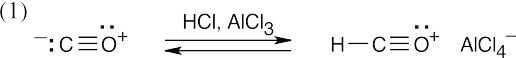
Carbon monoxide is protonated to form an acyl cation.
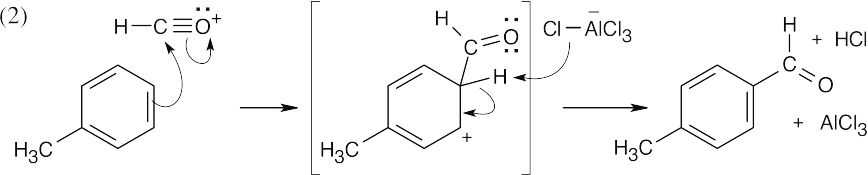
The acyl cation reacts with benzene by a Friedel–Crafts acylation mechanism. |
|
| 16.43 |
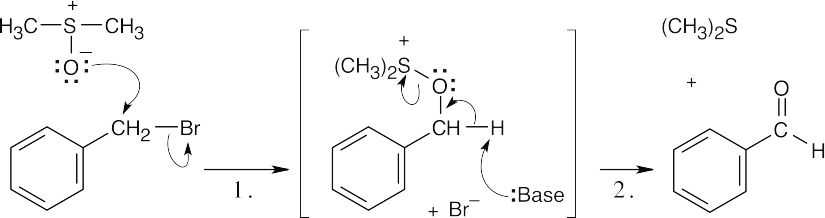
Step 1: SN2 displacement takes place when the negatively charged oxygen of dimethyl sulfoxide attacks the benzylic carbon of benzyl bromide, displacing Br–.Step 2: Base removes a benzylic proton, and dimethyl sulfide is eliminated in an E2 reaction. |
| 16.44 |
The Smiles Rearrangement is an intramolecular Nucleophilic Aromatic Substitution.
|
| 16.45 |
| (a)
Mechanism:
|
| (b)
Mechanism:
|
| (c)
Mechanism:
|
|
Reactivity and Orientation of Electrophilic Substitutions
| 16.47 |
Only methoxybenzene reacts faster than benzene (See Figure 16.12). |
| 16.48 |
Most reactive ———–> Least reactive
| (a) Benzene > Chlorobenzene > o-Dichlorobenzene |
| (b) Phenol > Nitrobenzene > p-Bromonitrobenzene |
| (c) o-Xylene > Fluorobenzene > Benzaldehyde |
| (d) p-Methoxybenzonitrile > p-Methylbenzonitrile > Benzonitrile |
|
| 16.49 |
| (a)
|
| (b)
Both groups direct substitution to the same position. |
| (c)

|
| (d)

|
| (e)
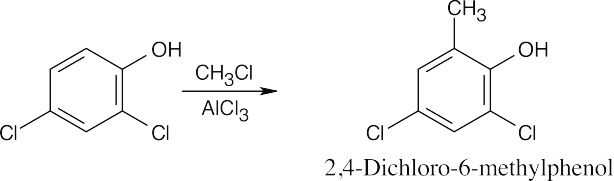
|
| (f)
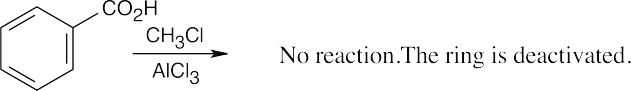
|
| (g)
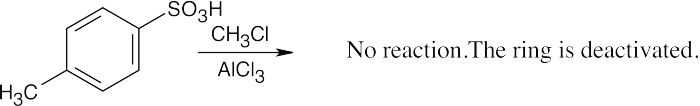
|
| (h)
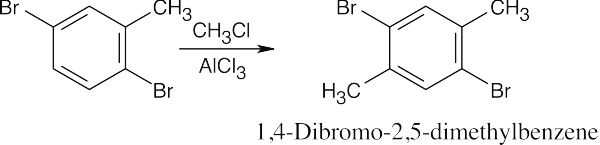
Alkylation occurs in the indicated position because the methyl group is more activating than bromine, and because substitution rarely takes place between two groups. |
|
| 16.50 |
| (a)
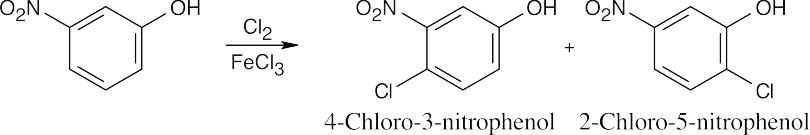
The –OH group directs the orientation of substitution. |
| (b)
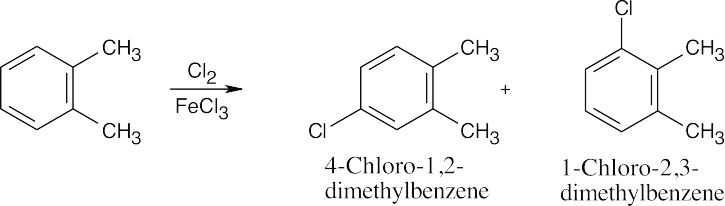
|
| (c)
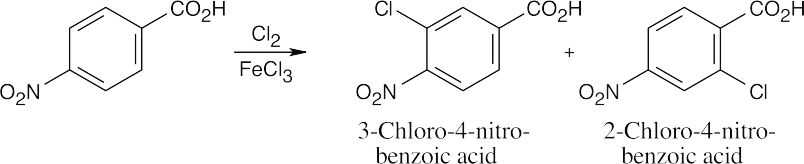
Both groups are deactivating to a similar extent, and both possible products form. |
| (d)
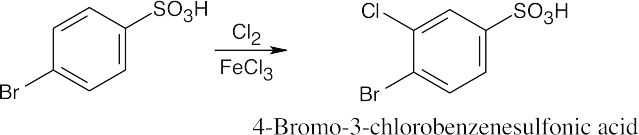
|
|
| 16.52 |
Most reactive ——–> Least reactive
Phenol > Toluene > p-Bromotoluene > Bromobenzene
Aniline and nitrobenzene don’t undergo Friedel–Crafts alkylations. |
| 16.53 |
| (a)
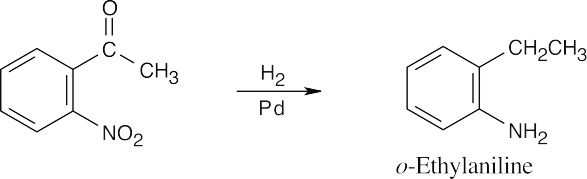
Catalytic hydrogenation reduces both the aromatic ketone and the nitro group. |
| (b)
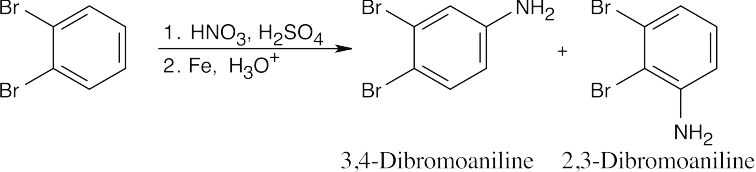
Nitration, followed by reduction with Fe, produces substituted anilines. |
| (c)
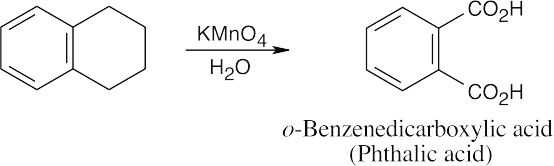
Aqueous KMnO4 oxidizes alkyl side chains to benzoic acids. |
| (d)
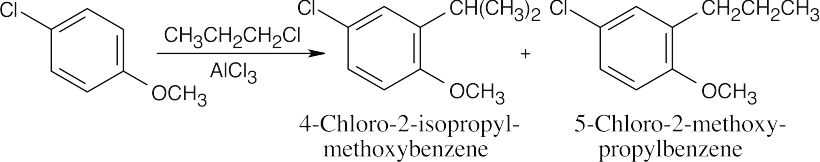
The methoxyl group directs substitution because it is a more powerful activating group. Rearranged and unrearranged side chains are present in the products. |
|
Organic Synthesis
| 16.55 |
| (a)
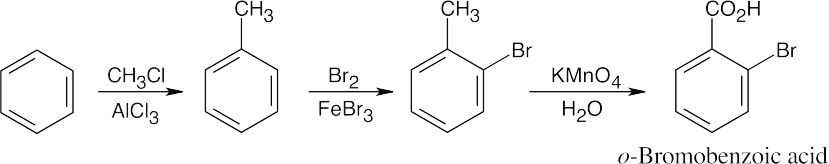
|
| (b)
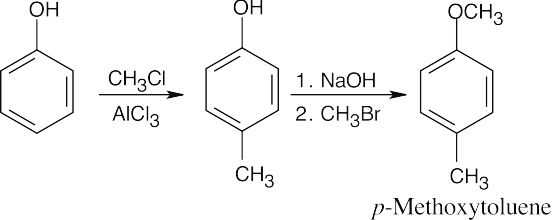
The reactions in (b) can be performed in either order. |
| (c)
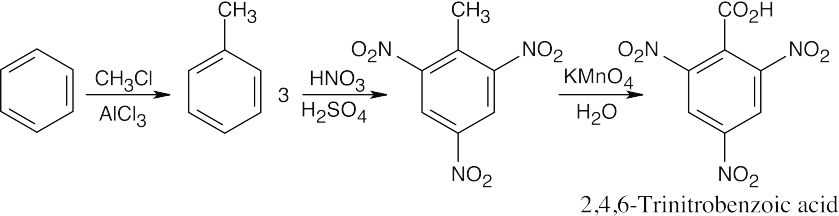
|
| (d)
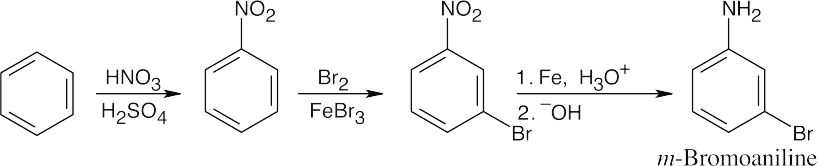
|
|
| 16.56 |
When synthesizing substituted aromatic rings, it is necessary to introduce substituents in the proper order. A group that is introduced out of order will not have the proper directing effect. Remember that in many of these reactions a mixture of ortho and para isomers may be formed.
|
| 16.57 |
| (a)
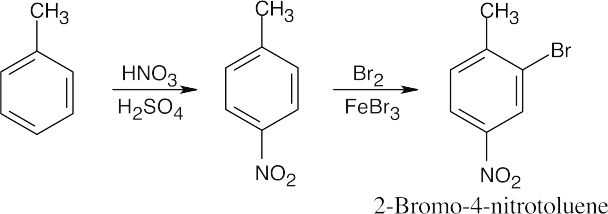
|
| (b)
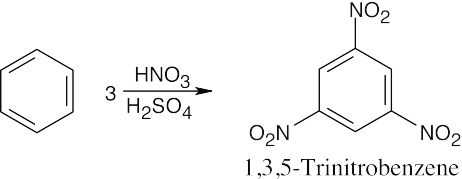
|
| (c)
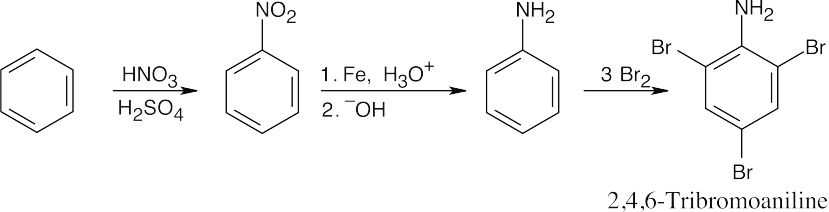
No catalyst is needed for bromination because aniline is very activated toward substitution. |
| (d)
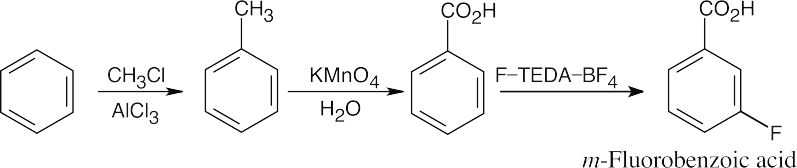
|
|
| 16.58 |
| (a) |
Chlorination of toluene occurs at the ortho and para positions. To synthesize the given product, first oxidize toluene to benzoic acid and then chlorinate. |
| (b) |
p-Chloronitrobenzene is inert to Friedel–Crafts alkylation because the ring is deactivated. |
| (c) |
The first two steps in the sequence are correct, but H2/Pd reduces the nitro group as well as the ketone. |
|
General Problems
| 16.59 |
Attack occurs on the unsubstituted ring because bromine is a deactivating group. Attack occurs at the ortho and para positions of the ring because the positively charged intermediate can be stabilized by resonance contributions from bromine and from the second ring (Problem 16.43).
|
| 16.60 |
When directly bonded to a ring, the –CN group is a meta-directing deactivator for both inductive and resonance reasons. In 3-phenylpropanenitrile, however, the saturated side chain does not allow resonance interactions of –CN with the aromatic ring, and the –CN group is too far from the ring for its inductive effect to be strongly felt. The side chain acts as an alkyl substituent, and ortho–para substitution is observed.
In 3-phenylpropenenitrile, the –CN group interacts with the ring through the π electrons of the side chain. Resonance forms show that –CN deactivates the ring toward electrophilic substitution, and substitution occurs at the meta position.
|
| 16.61 |
| (a)
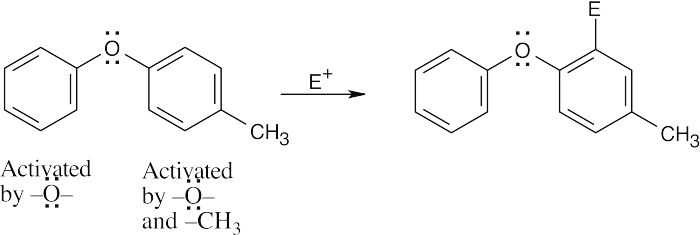
Substitution occurs in the more activated ring. The position of substitution is determined by the more powerful activating group – in this case, the ether oxygen. |
| (b)
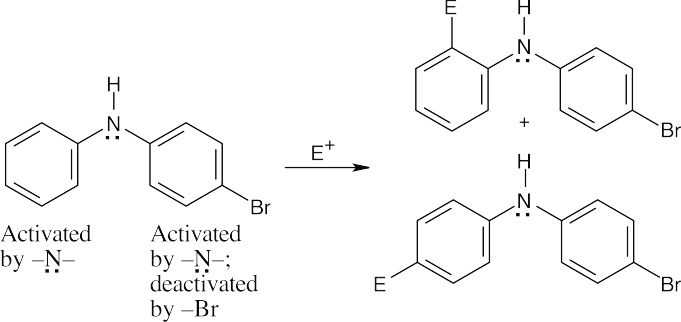
The left ring is more activated than the right ring. –NHR is an ortho-para director. |
| (c)
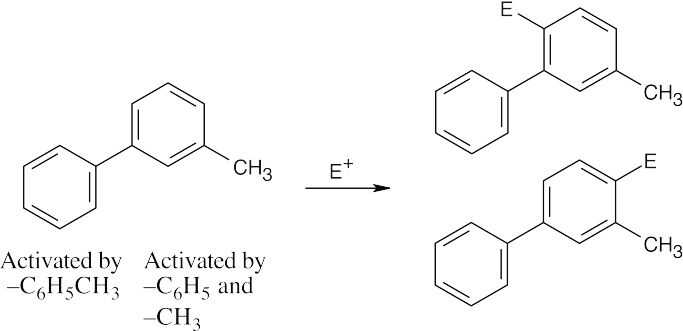
Substitution occurs at the ortho and para positions of the more activated ring. Substitution doesn’t occur between –C6H5 and –CH3 for steric reasons. |
| (d)
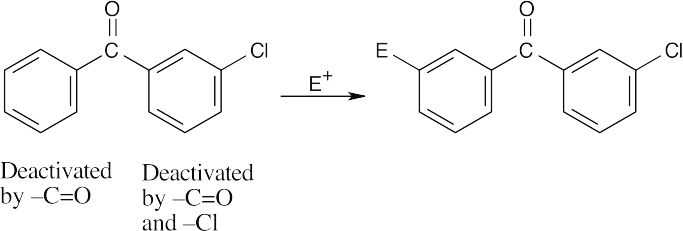
Substitution occurs at the meta positions of the ring on the left because it is less deactivated. |
|
| 16.62 |
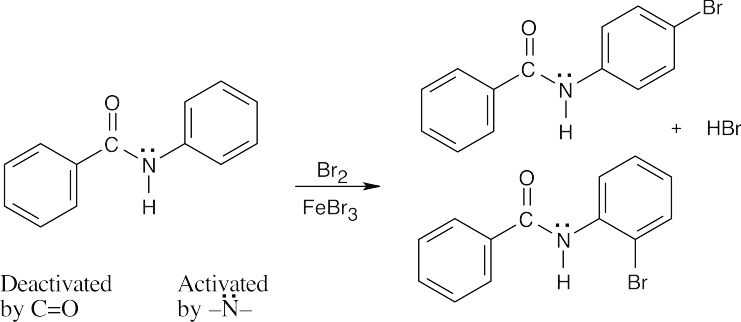
Attack occurs in the activated ring and yields ortho and para bromination products. The intermediate is resonance-stabilized by overlap of the nitrogen lone pair electrons with the π electrons of the substituted ring.
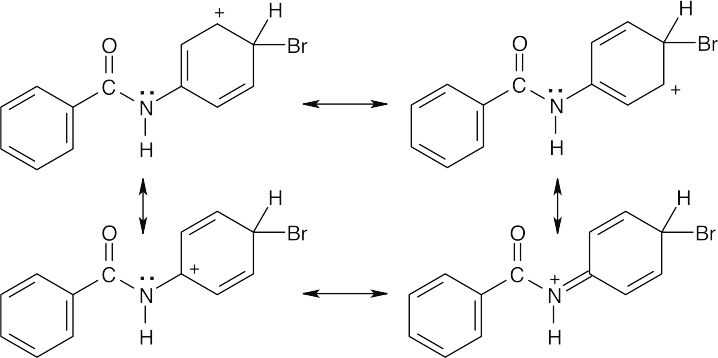
Similar drawings can be made of the resonance forms of the intermediate resulting from ortho attack. Even though the nitrogen lone-pair electrons are less available for delocalization than the lone-pair electrons of aniline (Problem 16.12), the –NH– group is nevertheless more activating than the C=O group. |
|
| 16.63 |
Reaction of (R)-2-chlorobutane with AlCl3 produces an ion pair [CH3+CHCH2CH3–AlCl4]. The planar, sp2-hybridized carbocation is achiral, and its reaction with benzene can occur on either side of the carbocation to yield racemic product. |
| 16.64 |
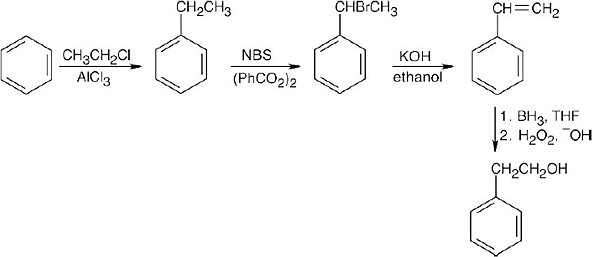
All of these syntheses involve NBS bromination of the benzylic position of a side chain.
|
| 16.65 |
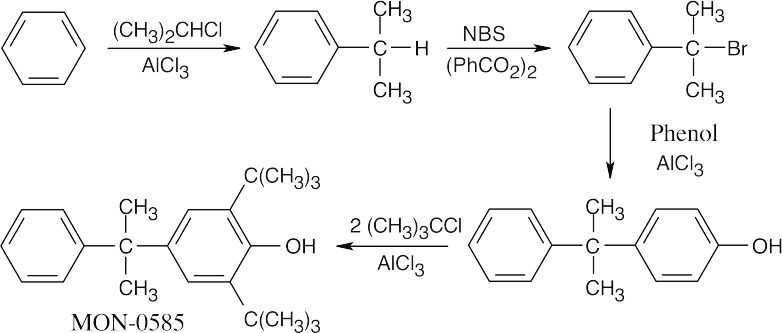
The product is a substituted phenol, whose –OH group directs the orientation of the –C(CH3)3 groups. The precursor to MON-0585 is synthesized by a Friedel–Crafts alkylation of phenol by the appropriate hydrocarbon halide. This compound is synthesized by NBS bromination of the product of alkylation of benzene with 2-chloropropane.
|
| 16.66 |
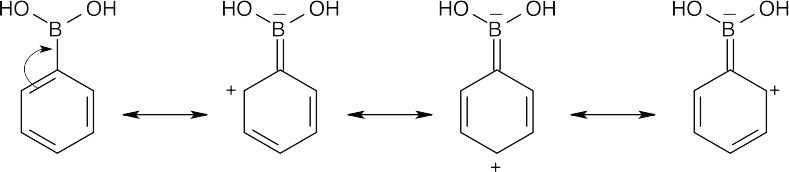
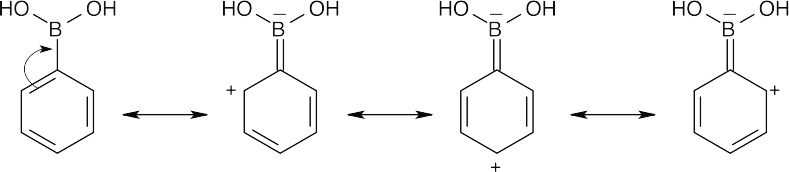
The trivalent boron atom in phenylboronic acid has only six outer-shell electrons and is a Lewis acid. It is possible to write resonance forms for phenylboronic acid in which an electron pair from the phenyl ring is delocalized onto boron. In these resonance forms, the ortho and para positions of phenylboronic acid are the most electron-deficient, and substitutions occur primarily at the meta position. |
| 16.67 |
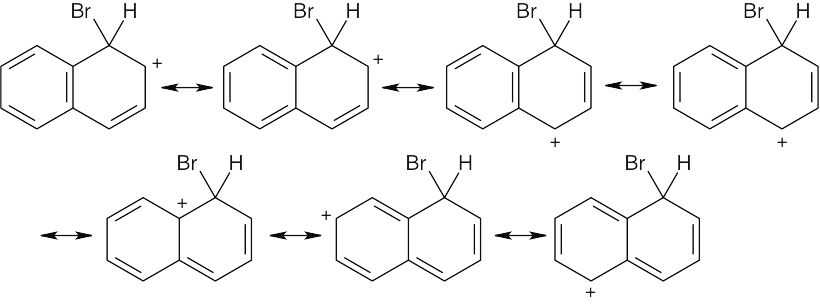
Resonance forms for the intermediate from attack at C1:
Resonance forms for the intermediate from attack at C2:
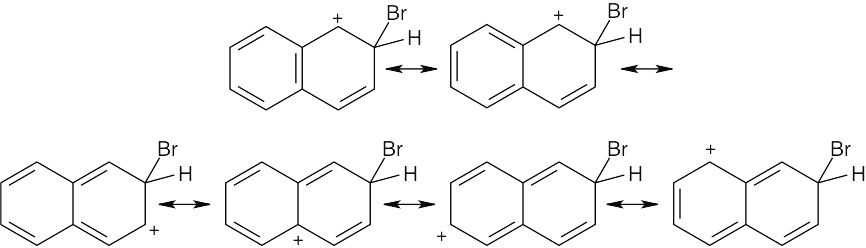
There are seven resonance forms for attack at C1 and six for attack at C2. Look carefully at the forms, however. In the first four resonance structures for C1 attack, the second ring is still fully aromatic. In the other three forms, however, the positive charge has been delocalized into the second ring, destroying the ring’s aromaticity. For C2 attack, only the first two resonance structures have a fully aromatic second ring. Since stabilization is lost when aromaticity is disrupted, the intermediate from C2 attack is less stable than the intermediate from C1 attack, and C1 attack is favored. |
|
| 16.68 |
Step 1: Addition of the nucleophile –OCH3.
Step 2: Elimination of –Cl–.
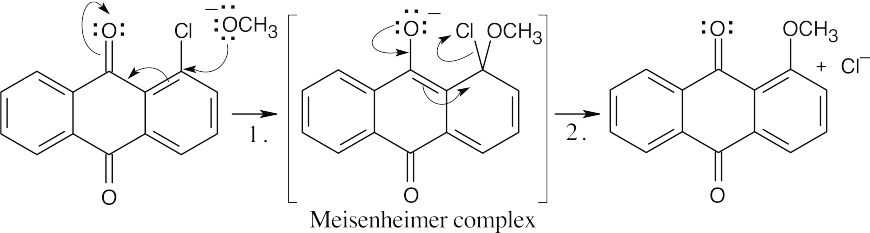
The carbonyl oxygens make the chlorine-containing ring electron-poor and open to attack by the nucleophile –OCH3. They also stabilize the negatively charged Meisenheimer complex. |
| 16.69 |
Step 1: Abstraction of proton and elimination of Br–.
Step 2: Addition of NH3 to the benzyne intermediate to form two aniline products. The reaction of an aryl halide with potassium amide proceeds through a benzyne intermediate. Ammonia can then add to either end of the triple bond to produce the two methylanilines observed. |
| 16.70 |
| (a)
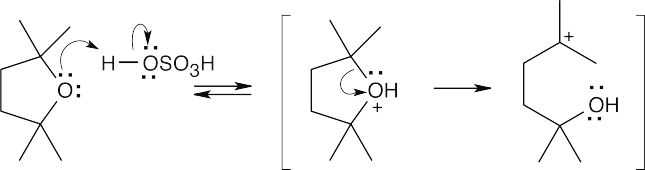
Protonation of the cyclic ether creates a carbocation intermediate that can react in a Friedel–Crafts alkylation. |
| (b)
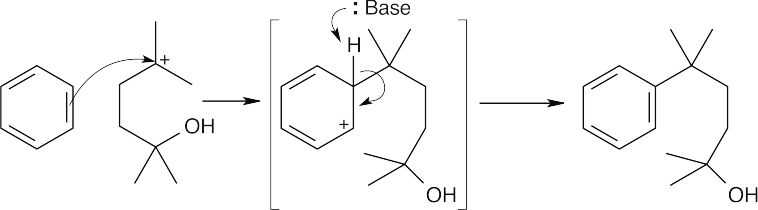
The intermediate alkylates benzene, forming an alcohol product. |
| (c)
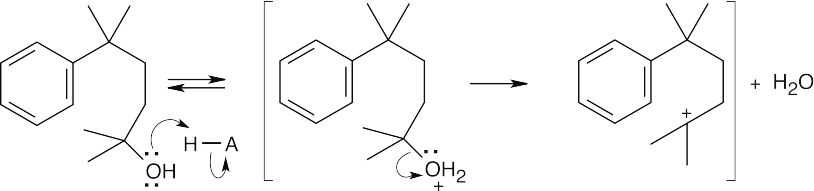
Protonation of the alcohol, followed by loss of water, generates a second carbocation. |
| (d)
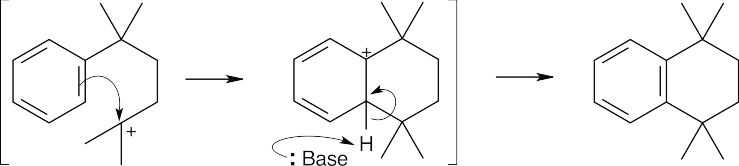
This carbocation undergoes internal alkylation to yield the observed product. |
|
| 16.71 |
Both of these syntheses test your ability to carry out steps in the correct order.
| (a)
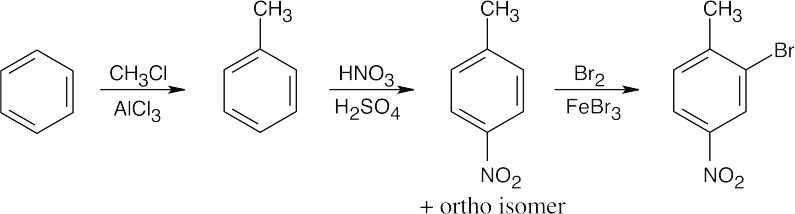
|
| (b)
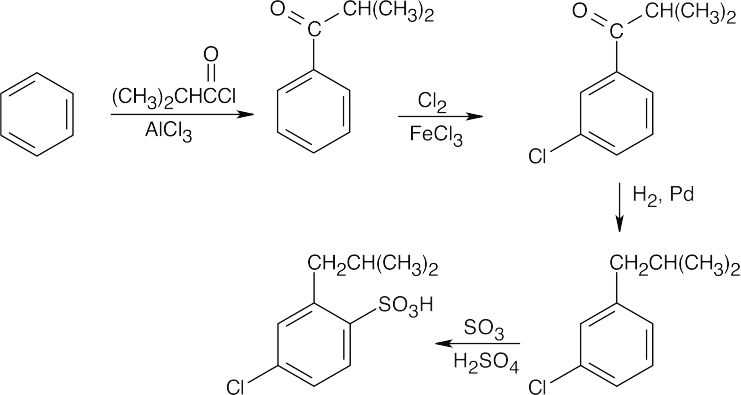
|
|
| 16.72 |
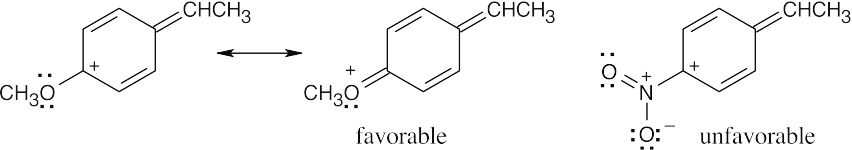
Problem 16.51 shows the mechanism of the addition of HBr to 1-phenylpropene and shows how the aromatic ring stabilizes the carbocation intermediate. For the methoxyl-substituted styrene, an additional resonance form can be drawn in which the cation is stabilized by the electron-donating resonance effect of the oxygen atom. For the nitro-substituted styrene, the cation is destabilized by the electron-withdrawing effect of the nitro group.
Thus, the intermediate resulting from addition of HBr to the methoxyl-substituted styrene is more stable, and reaction of p-methoxystyrene is faster. |
| 16.74 |
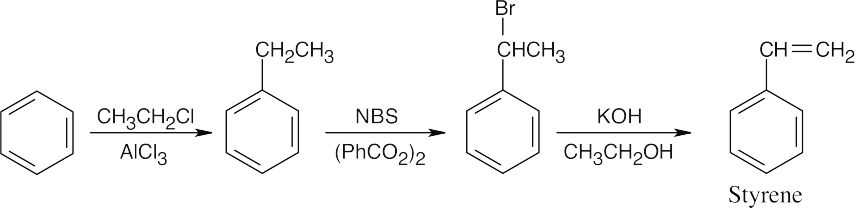
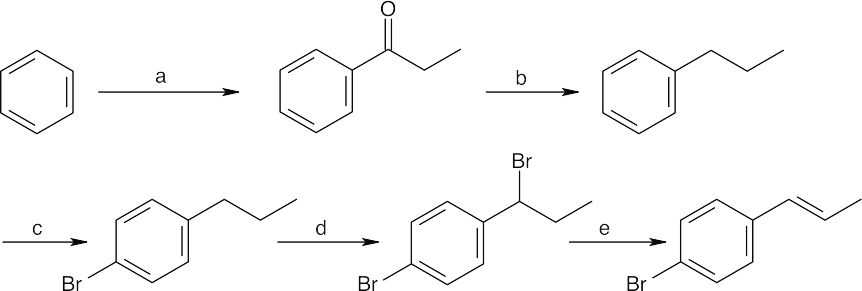
(a) CH3CH2COCl, AlCl3;
(b) H2, Pd/C;
(c) Br2, FeBr3;
(d) NBS, (PhCO2)2;
(e) KOH, ethanol |
| 16.75 |
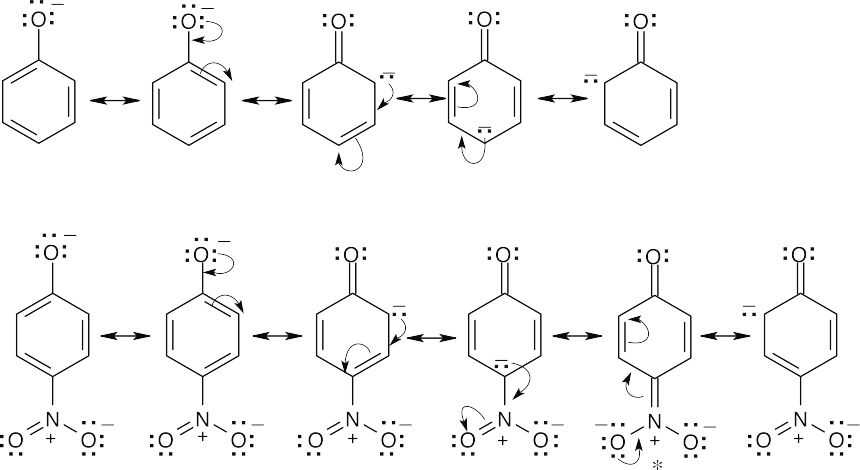
An electron-withdrawing substituent destabilizes a positively charged intermediate (as in electrophilic aromatic substitution) but stabilizes a negatively charged intermediate. For the dissociation of a phenol, an –NO2 group stabilizes the phenoxide anion by resonance, thus lowering ΔG° and pKa. In the starred resonance form for p-nitrophenol, the negative charge has been delocalized onto the oxygens of the nitro group. |
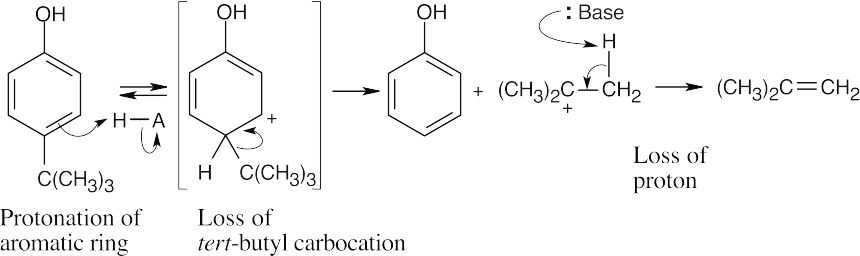
| 16.76 |
For the same reason described in the previous problem, a methyl group destabilizes the negatively charged intermediate, thus raising ΔG° and pKa, making this phenol less acidic. |
This file is copyright 2023, Rice University. All Rights Reserved.