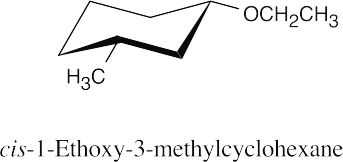
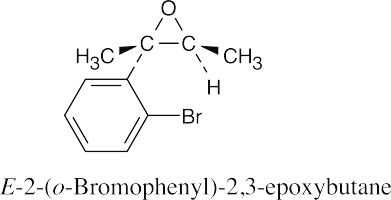
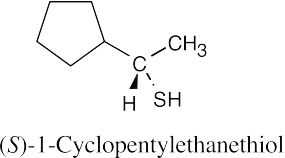
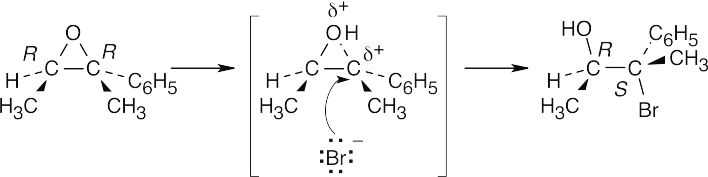
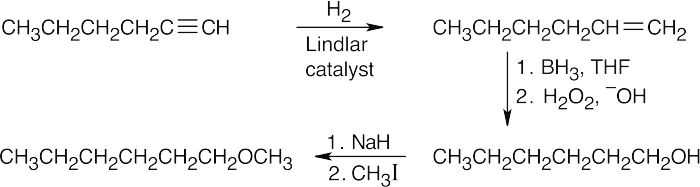
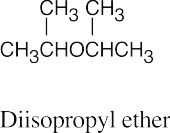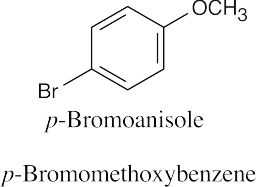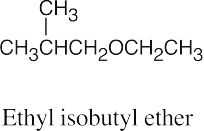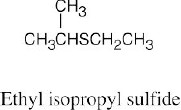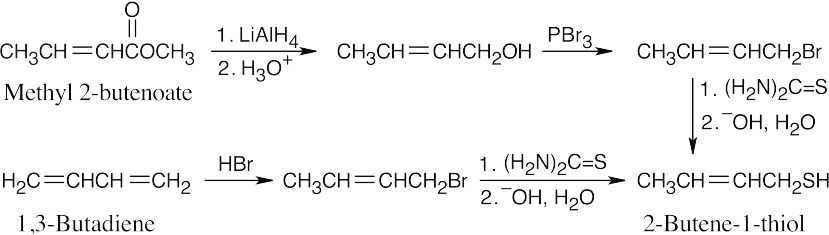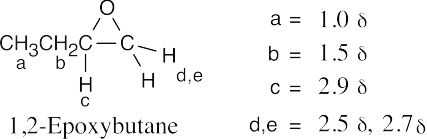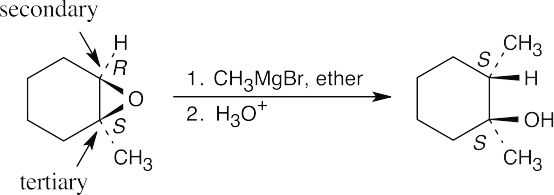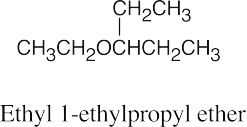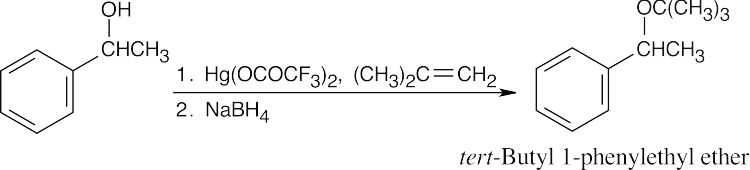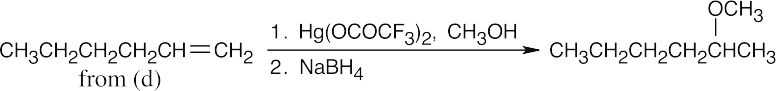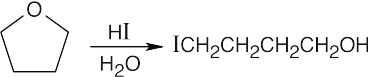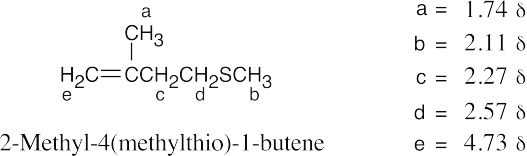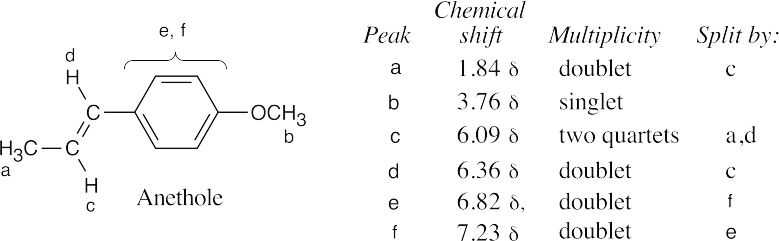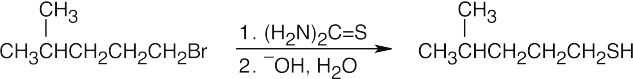18 Chapter 18 – Ethers and Epoxides; Thiols and Sulfides Solutions to Problems
Chapter 18 – Ethers and Epoxides; Thiols and Sulfides
Solutions to Problems
| 18.1 |
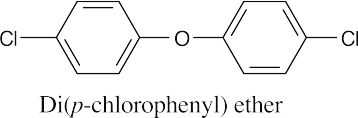
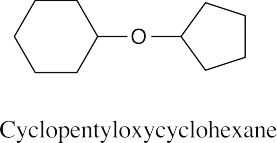
Ethers can be named either as alkoxy-substituted compounds or by citing the two groups bonded to oxygen, followed by the word “ether”.
|
| 18.2 |
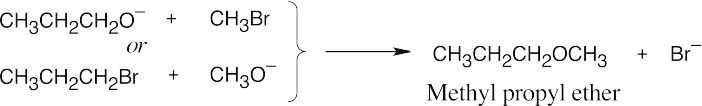
The first step of the dehydration mechanism is protonation of an alcohol. Water is then displaced by another molecule of alcohol to form an ether. If two different alcohols are present, either one can be protonated and either one can displace water, yielding a mixture of products.
If this procedure were used with ethanol and 1-propanol, the products would be diethyl ether, ethyl propyl ether, and dipropyl ether. If there were equimolar amounts of the alcohols, and if they were of equal reactivity, the product ratio would be diethyl ether: ethyl propyl ether: dipropyl ether = 1:2:1. |
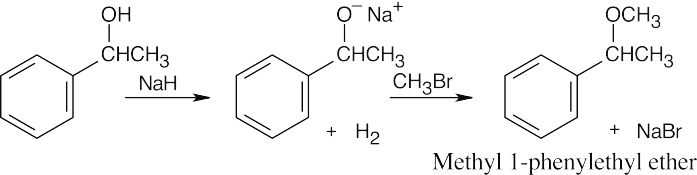
| 18.3 |
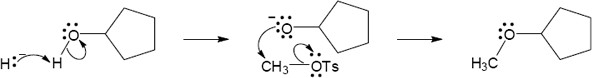
Remember that the halide in the Williamson ether synthesis should be primary or methyl, in order to avoid competing elimination reactions. The alkoxide anions shown are formed by treating the corresponding alcohols with NaH.
|
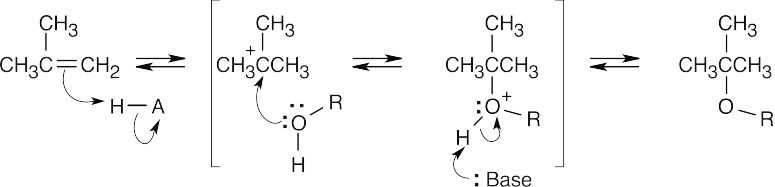
| 18.4 |
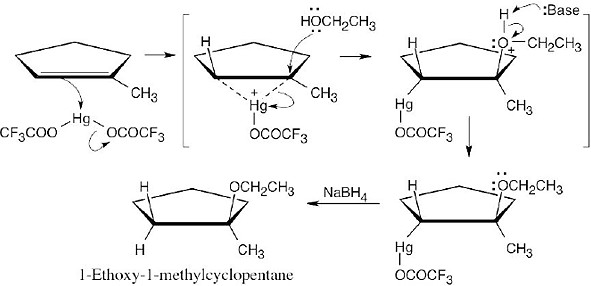
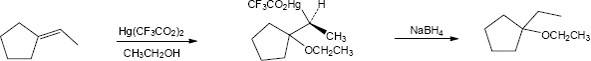
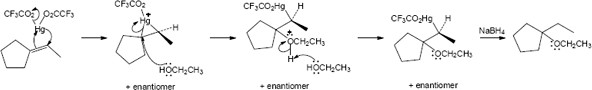
The reaction mechanism of alkoxymercuration/demercuration of an alkene is similar to other electrophilic additions we have studied. First, the cyclopentene π electrons attack Hg2+ with formation of a mercurinium ion. Next, the nucleophilic alcohol displaces mercury. Markovnikov addition occurs because the carbon bearing the methyl group is better able to stabilize the partial positive charge arising from cleavage of the carbon-mercury bond. The ethoxyl and mercuric groups are trans to each other. Finally, removal of mercury by NaBH4 by a mechanism that is not fully understood results in the formation of 1-ethoxy-1-methylcyclopentane. |
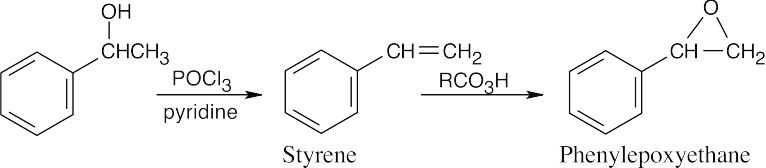
| 18.5 |
Use the Williamson synthesis when one of the ether components can be a primary or benzylic halide. Use alkoxymercuration when one or both components are branched.
| (a) Either method of synthesis is appropriate.
Williamson:
Alkoxymercuration:
|
| (b) Either method is possible, but the Williamson synthesis is simpler.
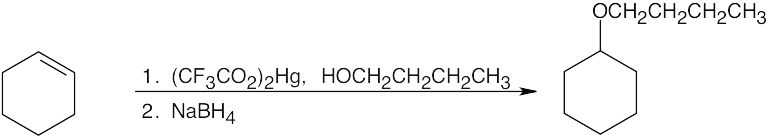
|
| (c) Use alkoxymercuration because both parts of the ether are branched.
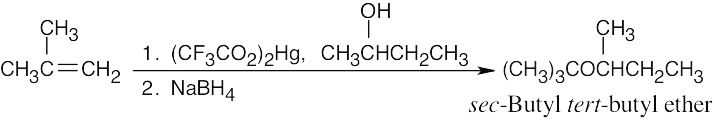
|
| (d) The Williamson synthesis must be used.
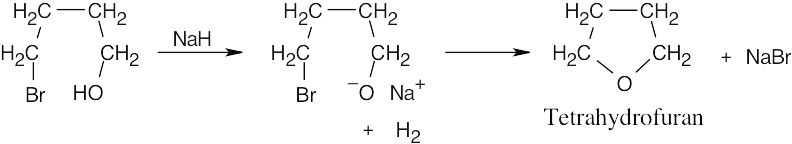
|
|
| 18.6 |
The compounds most reactive in the Williamson ether synthesis are also most reactive in any SN2 reaction (review Chapter 11 if necessary).
Most reactive ———–> Least reactive
| (a)
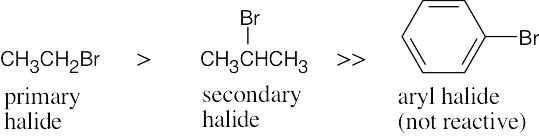
|
| (b)
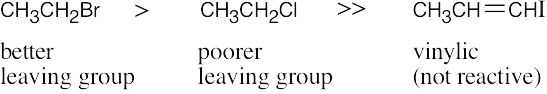
|
|
| 18.7 |
| (a) |
First, notice the substitution pattern of the ether. Bonded to the ether oxygen are a primary alkyl group and a tertiary alkyl group. When one group is tertiary, cleavage occurs by an SN1 or E1 route to give either an alkene or a tertiary halide and a primary alcohol.
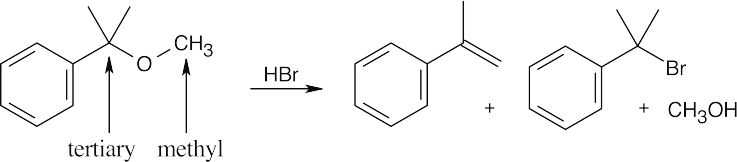
|
| (b) |
In this problem, the groups are primary and secondary alkyl groups. Br– attacks at the less hindered primary group, and oxygen remains with the secondary group, to give a secondary alcohol.
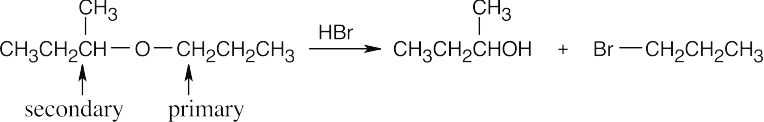
|
|
| 18.8 |
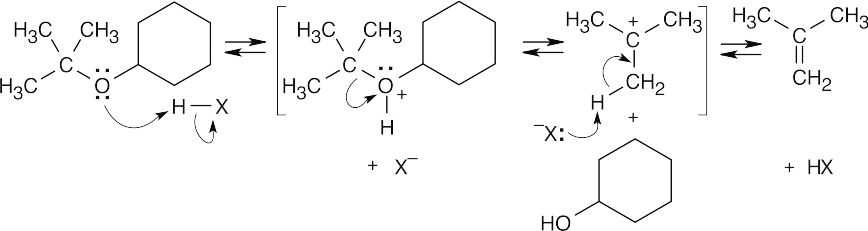
The first step of acid-catalyzed ether cleavage is protonation of the ether oxygen to give an intermediate oxonium ion, which collapses to form an alcohol and a tertiary carbocation. The carbocation then loses a proton to form an alkene, 2-methylpropene. This is an example of E1 elimination. The acid used for cleavage is often trifluoroacetic acid. |
| 18.9 |
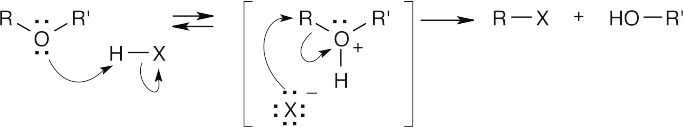
HX first protonates the oxygen atom, and halide then brings about a nucleophilic displacement to form an alcohol and an organic halide. The better the nucleophile, the more effective the displacement. Since I– and Br– are better nucleophiles than Cl–, ether cleavage proceeds more smoothly with HI or HBr than with HCl. |
| 18.10 |
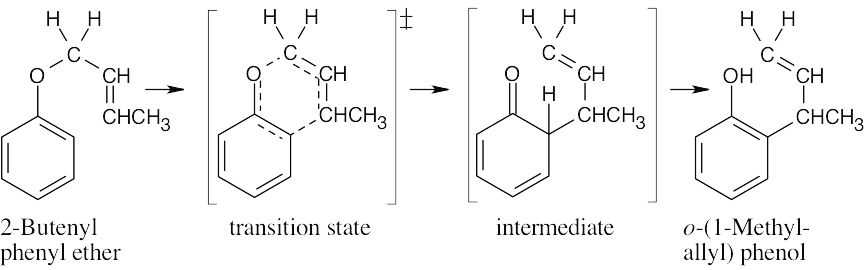
Draw the ether with the groups involved in the rearrangement positioned as they will appear in the product. Six bonds will either be broken or formed in the product; they are shown as dashed lines in the transition state. Redraw the bonds to arrive at the intermediate enone, which rearranges to the more stable phenol.
|
| 18.11 |
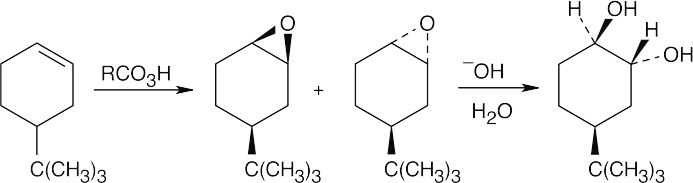
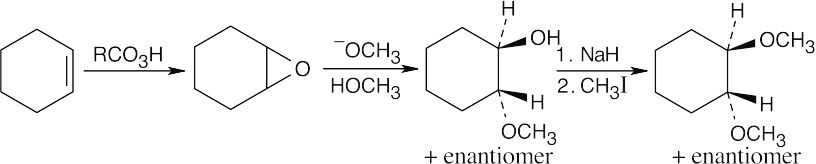
Epoxidation by use of m-chloroperoxybenzoic acid (RCO3H) is a syn addition of oxygen to a double bond. The original bond stereochemistry is retained, and the product is a meso compound.
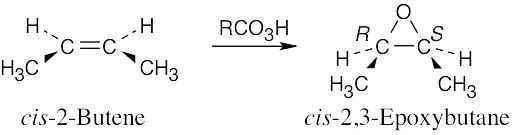
In the epoxide product, as in the alkene starting material, the methyl groups are cis.
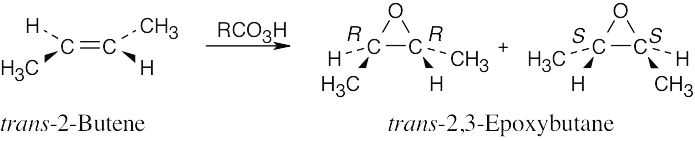
Reaction of trans-2-butene with m-chloroperoxybenzoic acid yields trans-2,3- epoxybutane. A mixture of enantiomers is formed because the peroxyacid can attack either the top or bottom of the double bond. |
|
| 18.12 |
As discussed in this section, acid-catalyzed epoxide ring opening occurs primarily at the more hindered carbon if one of the epoxide carbons is tertiary. In both parts of this problem, one epoxide carbon is tertiary.
| (a)
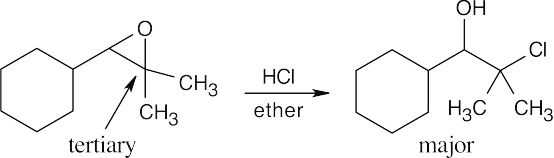
|
| (b)
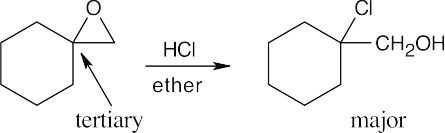
|
|
| 18.13 |
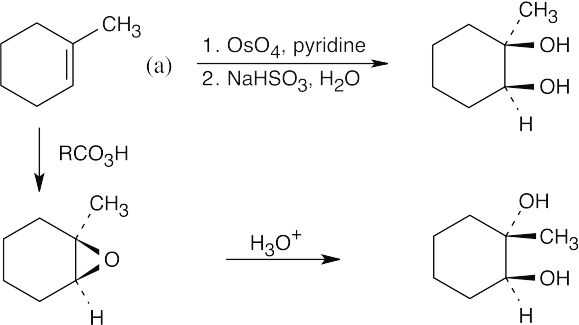
Notice the relationship of the hydroxyl groups in the two diols. In diol (a), the two hydroxyls are cis, and in (b) they are trans. Since ring-opening of epoxides forms trans-1,2-diols, only diol (b) can be formed by this route. The cis-1,2-diol in (a), results from treatment of 1-methylcyclohexene with OsO4. The enantiomers of the diols are also formed.
|
| 18.14 |
| (a) |
Attack of the basic nucleophile occurs at the less substituted epoxide carbon.
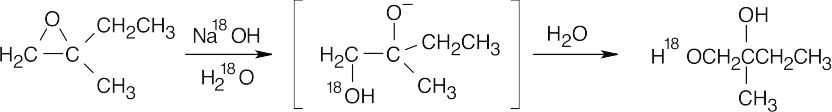
|
| (b) |
Under acidic conditions, ring-opening occurs at the more substituted epoxide carbon when one of the carbons is tertiary.
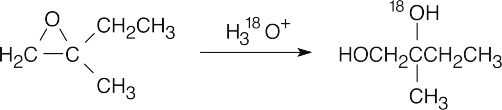
|
| (c) |
Addition of a Grignard reagent takes place at the less substituted epoxide carbon.
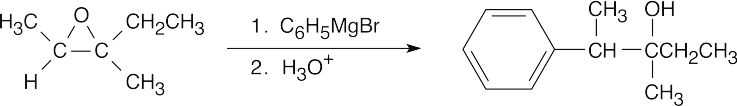
|
|
| 18.15 |
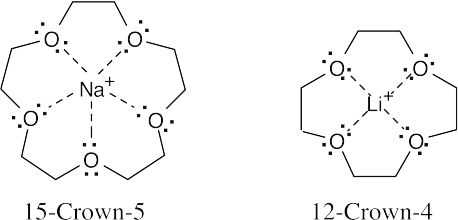
Bases on ionic radii, the ion-to-oxygen distance in 15-crown-5 is about 40% longer than the ion-to-oxygen distance in 12-crown-4. |
| 18.16 |
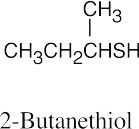
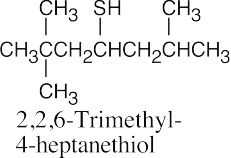
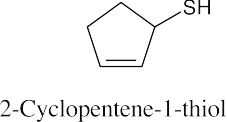
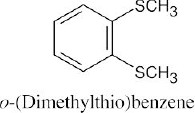
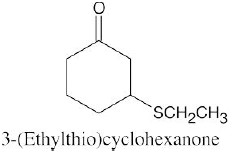
Thiols are named by the same rules as alcohols, with the suffix -ol replaced by the suffix-thiol. Sulfides are named by the same rules as ethers, with “sulfide” replacing “ether”.
|
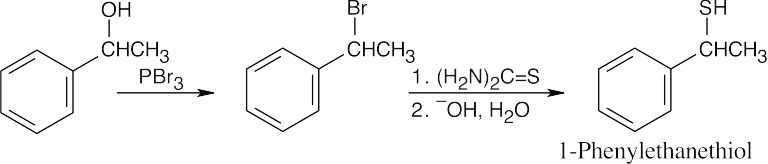
| 18.17 |
Thiourea is used to prepare thiols from alkyl halides.
|
Additional Problems
Visualizing Chemistry
| 18.20 |
Ring-opening occurs at the tertiary carbon to give carbocation-like stability to the transition state. Bromine approaches 180° from the C–OH bond, as it would in an SN2 reaction.
|
| 18.21 |
The Grignard reagent attacks the epoxide at the less hindered carbon in an SN2 reaction. The oxygen remains bonded to the tertiary carbon.
|
| 18.22 |
A molecular model shows that approach to the upper face of the double bond is hindered by a methyl group. Reaction with RCO3H occurs at the lower face of the double bond to produce epoxide A.
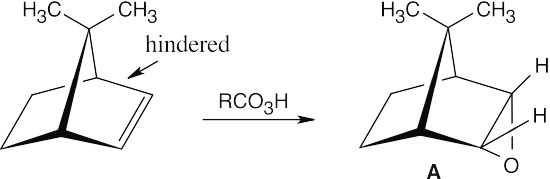
In the reaction of Br2 and H2O, the intermediate bromonium ion also forms at the lower face. Reaction with water yields a bromohydrin which, when treated with a base, forms epoxide B.
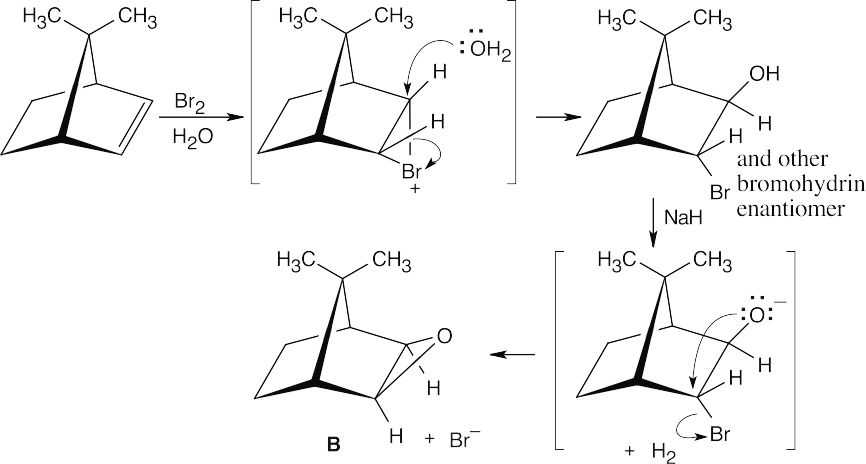
|
|
Mechanism Problems
| 18.23 |
| (a)
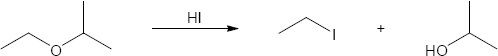
Mechanism:
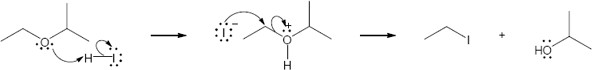
|
| (b)
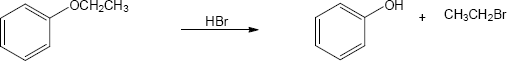
Mechanism:
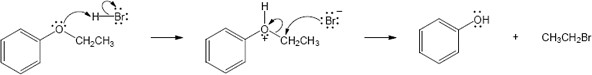
|
|
| 18.24 |
| (a)
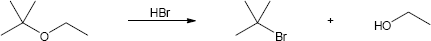
Mechanism:

|
| (b)
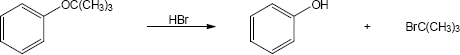
Mechanism:
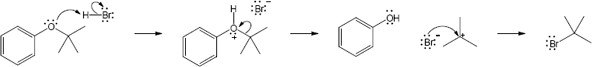
|
|
| 18.25 |
| (a)
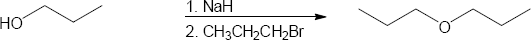
Mechanism:

|
| (b)
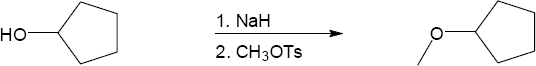
Mechanism:
|
|
| 18.26 |
Mechanism:
|
|
| 18.27 |
|
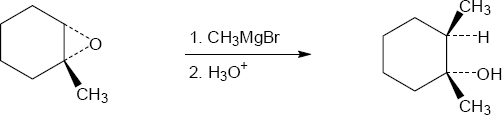
Mechanism:
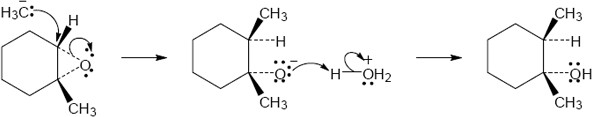
|
|
|
|
| 18.28 |
| (a)
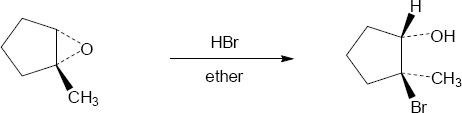
Mechanism:
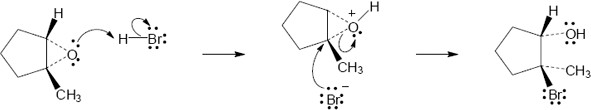
|
| (b)
Mechanism:
|
|
| 18.29 |
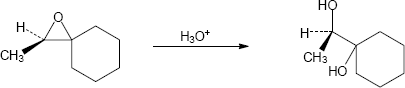
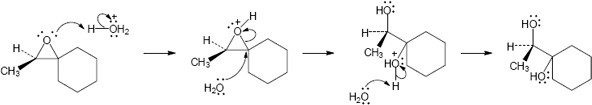
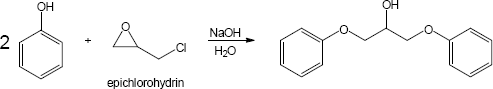
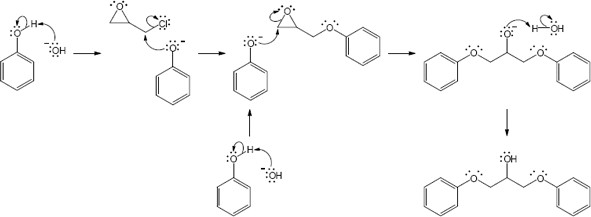
Note: The phenolate ion serves as a nucleophile both to displace chloride ion and to open the epoxide. While the mechanism here shows the displacement of chloride first, it is likely that the rate of epoxide opening is competitive. Thus, either epoxide opening or chloride opening may be first. In either case the final product is the same.
Mechanism:
|
|
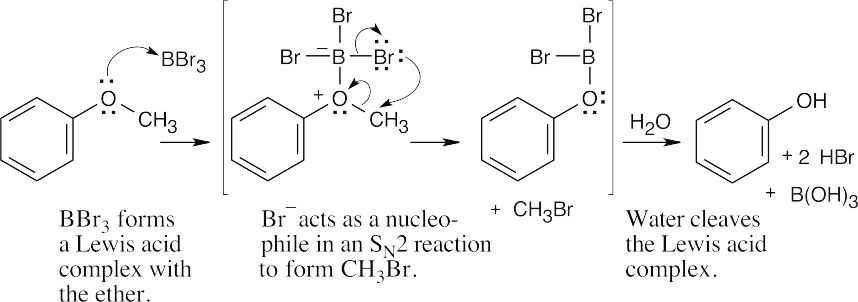
| 18.30 |
Step 1: BBr3 forms a Lewis acid complex with the ether.
Step 2: Br– acts as a nucleophile in an SN2 reaction to form CH3Br.
Step 3: Water cleaves the Lewis acid complex. |
| 18.31 |
Step 1: Protonation.
Step 2: Epoxide opening.
Step 3: Hydride shift.
Step 4: Loss of proton.
Reaction occurs by this route because of the stability of the intermediate tertiary carbocation. |
| 18.32 |
| (a) |
This is an SN2 reaction because the rate depends on the concentrations of both reagents. |
| (b) |
This SN2 reaction is a Williamson ether synthesis, in which an alkoxide displaces a halogen. In this reaction, KOH is used to form the phenoxide anion.
|
|
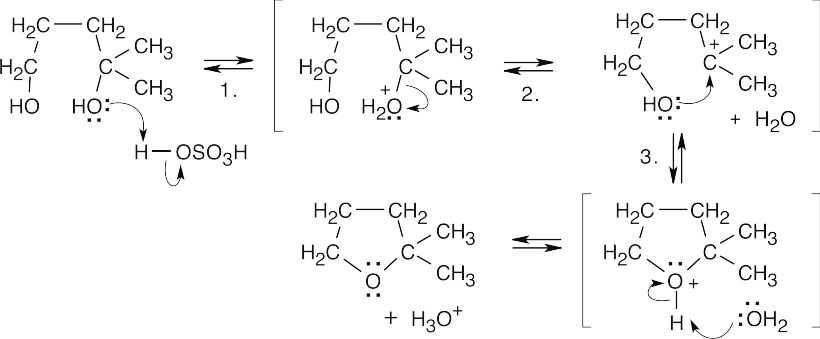
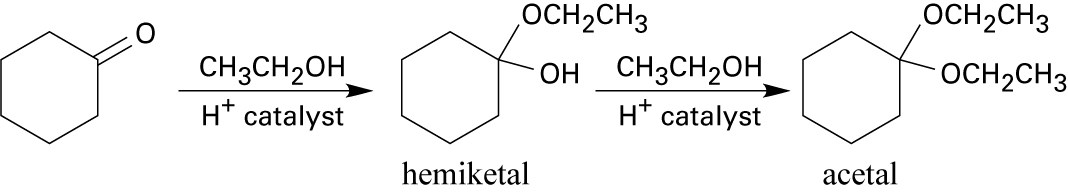
| 18.33 |
Step 1: Protonation of the tertiary hydroxyl group.
Step 2: Loss of water to form a tertiary carbocation.
Step 3: Nucleophilic attack on the carbocation by the second hydroxyl group.
The tertiary hydroxyl group is more likely to be eliminated because the resulting carbocation is more stable. |
| 18.34 |
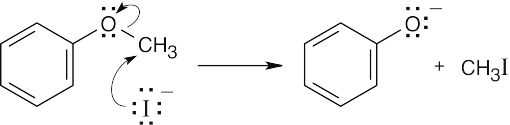
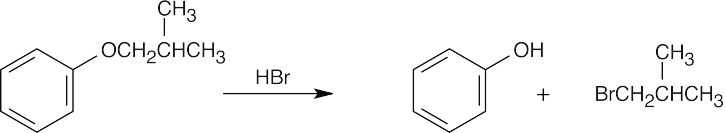
This reaction is an SN2 displacement and can’t occur at an aryl carbon. DMF is a polar aprotic solvent that increases the rate of an SN2 reaction by making anions more nucleophilic. |
| 18.35 |
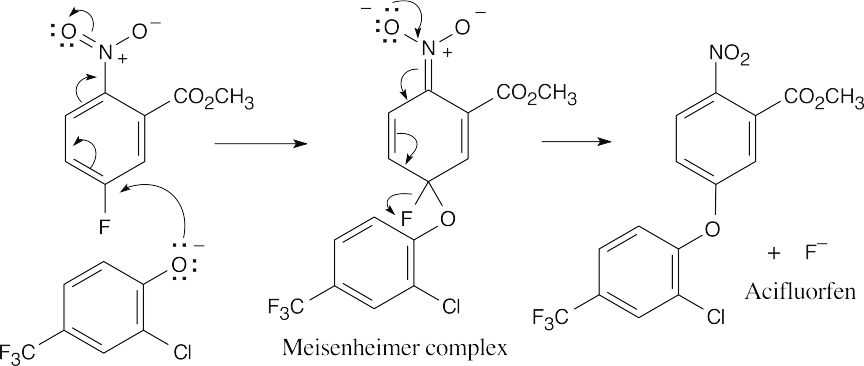
The reaction is a nucleophilic aromatic substitution. The intermediate Meisenheimer complex is stabilized by the –NO2 group.
Step 1: Addition of phenoxide.
Step 2: Elimination of fluoride. |
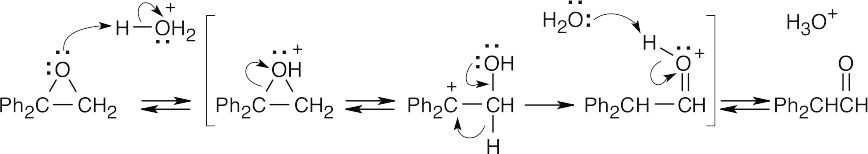
| 18.36 |
| (a)
|
| (b)
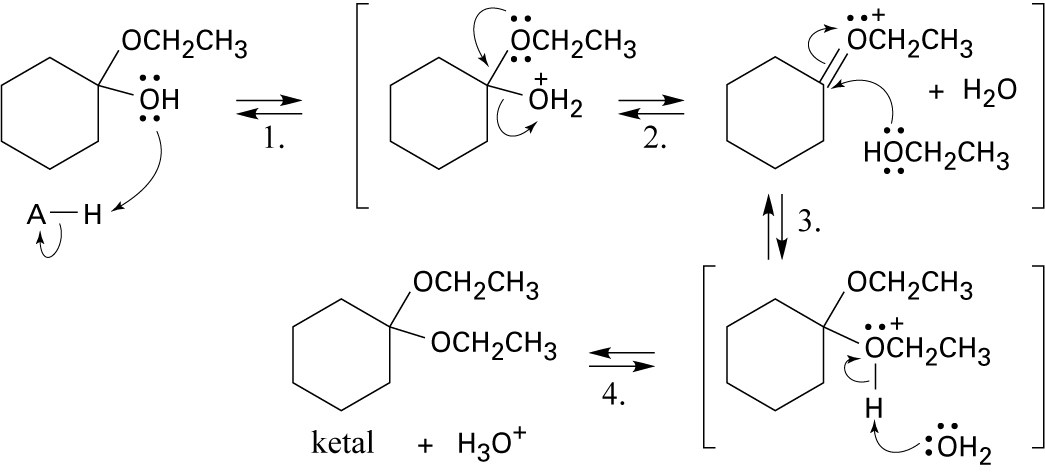
|
Step 1: Protonation.
Step 2: Loss of water.
Step 3: Addition of ethanol.
Step 4: Loss of proton. |
| 18.37 |
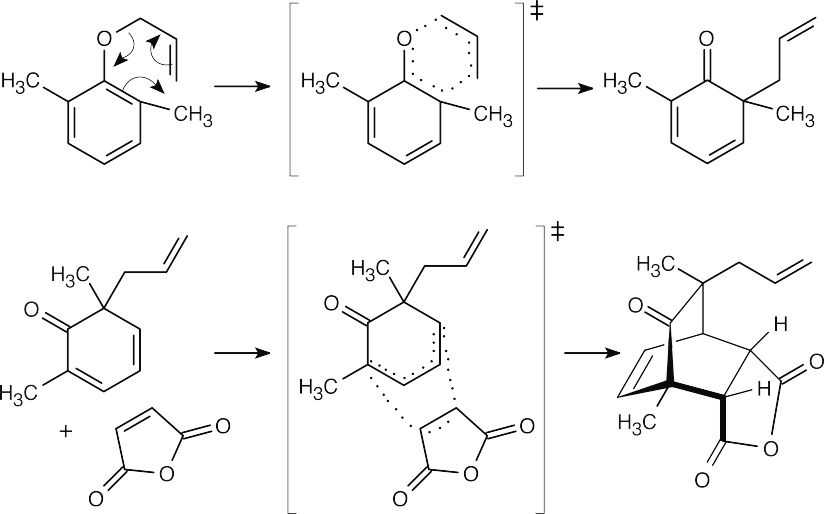
A Claisen rearrangement is followed by a Diels–Alder reaction (Section 14.4). Tautomerization of the Claisen product to a phenol doesn’t take place because no hydrogen is available for donation to oxygen.
|
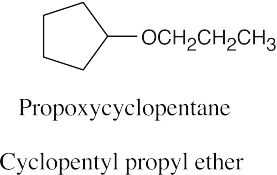
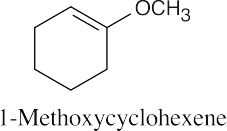
Naming Ethers
| 18.39 |
| (a)
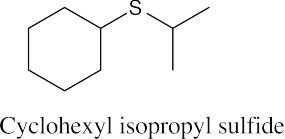
|
(b)
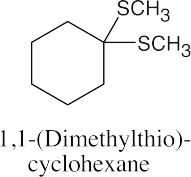
|
| (c)
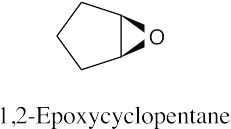
|
(d)
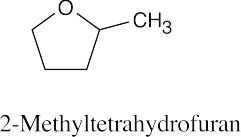
|
| (e)
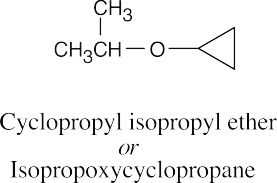
|
(f)
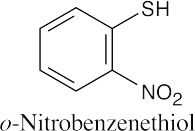
|
| (g)
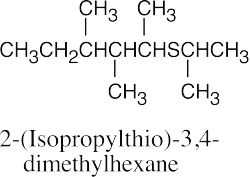
|
(h)
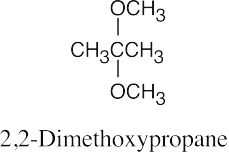
|
| (i)
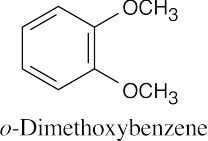
|
|
|
Synthesizing Ethers
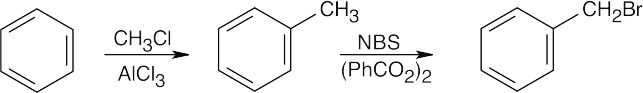
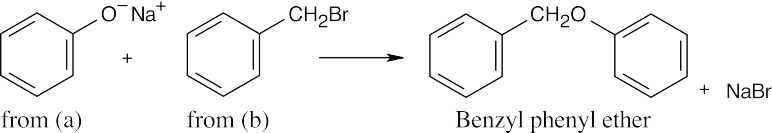
| 18.40 |
| (a)
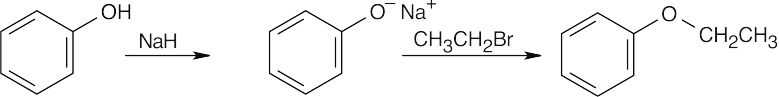
|
| (b)
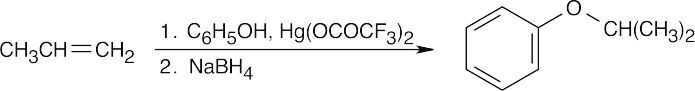
|
| (c)
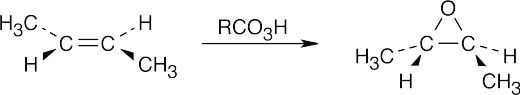
[RCO3H = meta-Chloroperoxybenzoic acid] |
| (d)
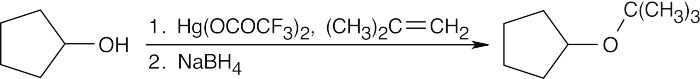
|
| (e)
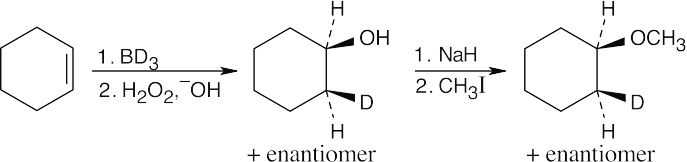
|
| (f)
|
|
| 18.42 |
Step 1: Protonation.
Step 2: Attack of alcohol oxygen on carbocation.
Step 3: Loss of proton.
Notice that this reaction is the reverse of acid-catalyzed cleavage of a tertiary ether (Problem 18.8). |
| 18.43 |
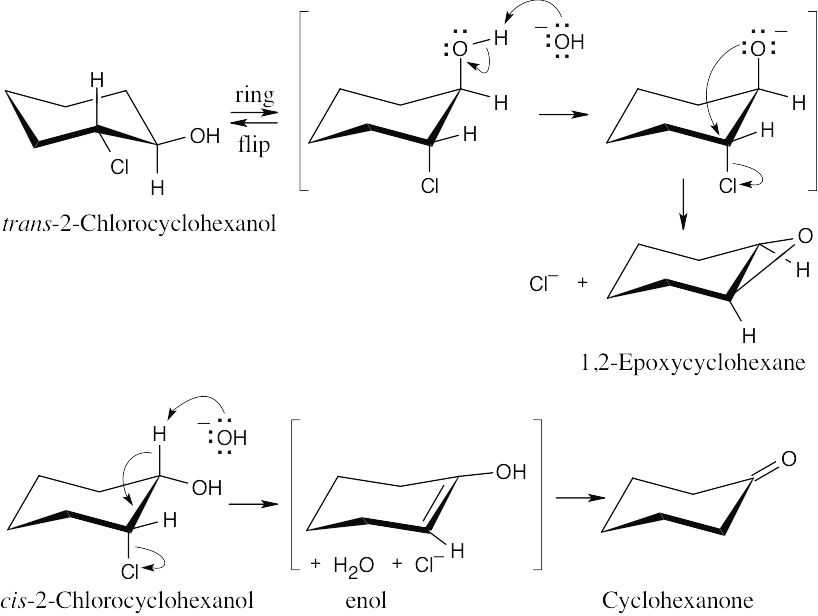
In the trans isomer, the –OH and –Cl are in the trans orientation that allows epoxide formation to occur as described in Section 18.4. Epoxidation can’t occur for the cis isomer, however. Instead, the base –OH brings about E2 elimination, producing an enol, which tautomerizes to cyclohexanone.
|
Reactions of Ethers and Epoxides
| 18.44 |
| (a)
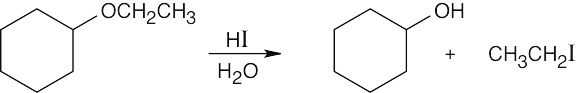
|
| (b)
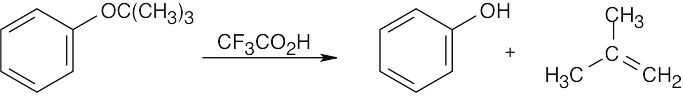
|
| (c)
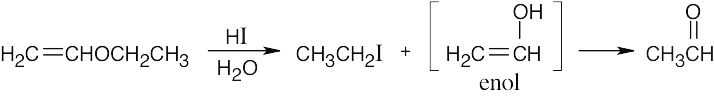
The enol tautomerizes to an aldehyde. |
| (d)
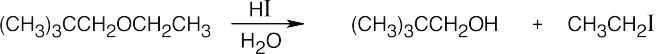
|
|
| 18.47 |
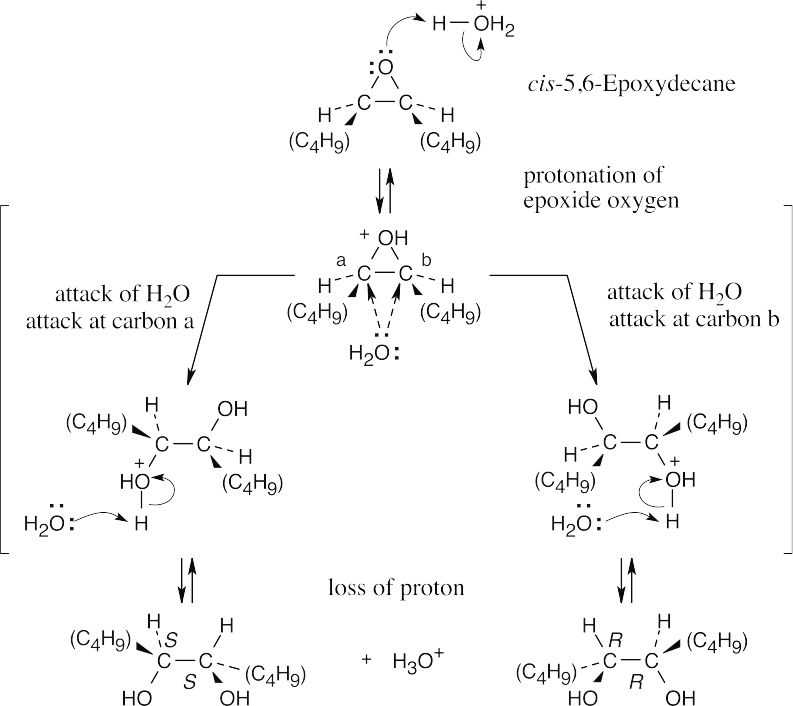
The product of acid hydrolysis of cis-5,6-epoxydecane is a racemic mixture of R,R and S,S diols. |
| 18.48 |
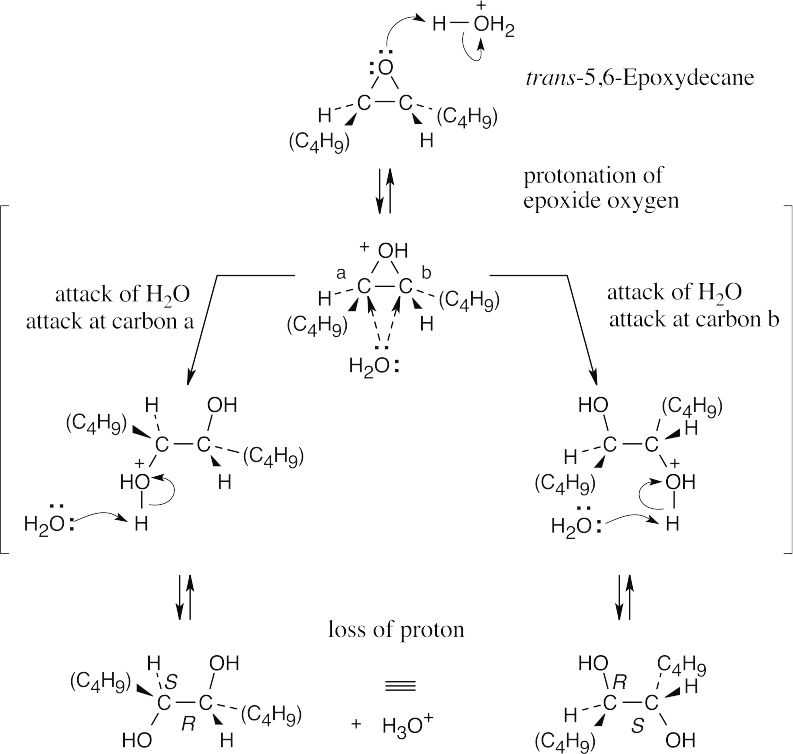
The product of acid hydrolysis of trans-5,6-epoxydecane is a meso compound that is a diastereomer of the products formed in the previous problem. |
| 18.49 |
The hydroxyl groups in the product have a trans-diaxial relationship. |
| 18.50 |
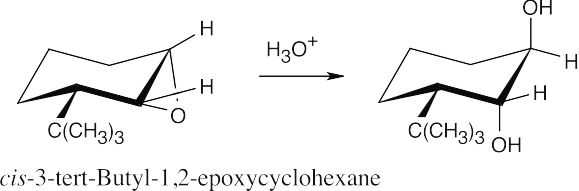
| (a) (b) |
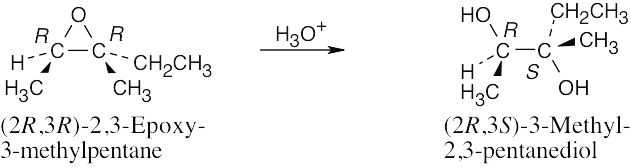
Reaction with aqueous acid causes ring opening to occur at C3 because the positive charge of the transition state is more stabilized at the tertiary carbon. Ring opening produces a diol in which the hydroxyl groups have a trans-diaxial relationship. |
| (c) |
Since ring opening occurs exclusively at C3, the product is the 2R,3S isomer and is chiral. (If ring opening occurred equally at either carbon, the product would be a mixture of chiral enantiomers). |
| (d) |
The product is optically active because only one enantiomer is produced. |
|
| 18.51 |
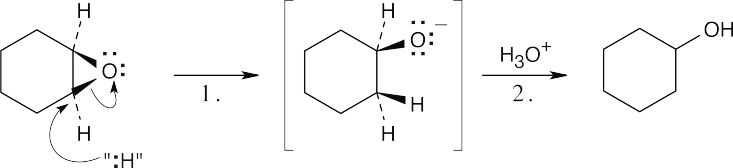
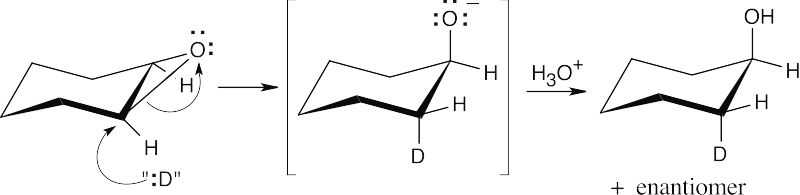
Step 1: Attack of the hydride nucleophile.
Step 2: Protonation of the alkoxide anion.
The reaction is an SN2 epoxide cleavage with “:H” as the nucleophile. The exact nature of the attacking nucleophile is not clear. |
| 18.52 |
Deuterium and –OH have a trans-diaxial relationship in the product. |
Spectroscopy
| 18.53 |
M+ = 116 corresponds to a sulfide of molecular formula C6H12S, indicating one degree of unsaturation. The IR absorption at 890 cm–1 is due to a R2C=CH2 group.
|
| 18.55 |
| (a)
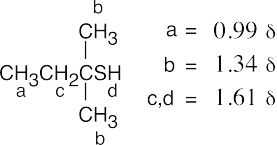
|
(b)
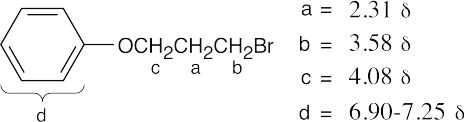
|
| (c)
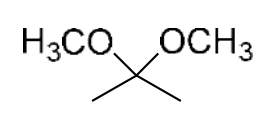
2,2-Dimethoxypropane |
|
|
General Problems
| 18.57 |
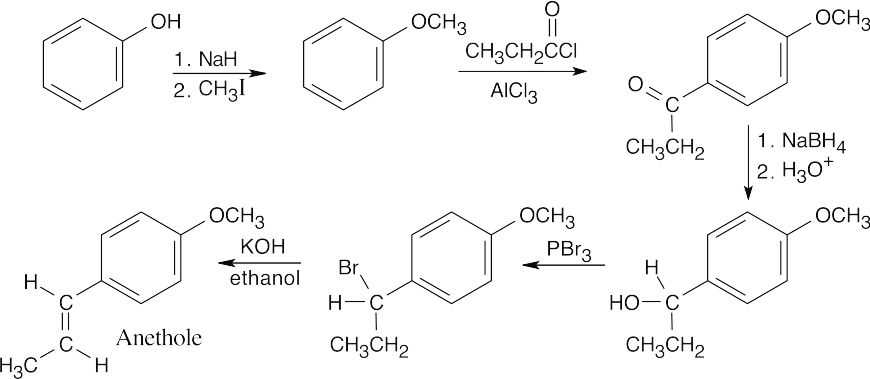
The anethole ring has two functional groups – an ether and a hydrocarbon side chain with a double bond. The ether is synthesized first – by a Williamson ether synthesis from phenol and CH3I. The hydrocarbon side chain results from a Friedel–Crafts acylation of the ether. Reduction of the ketone, bromination and dehydrohalogenation are used to introduce the double bond. |
| 18.59 |
Step 1: Attack of the alcohol on the triethyloxonium cation, with loss of diethyl ether.
Step 2: Loss of proton.
Trialkyloxonium salts are more reactive alkylating agents than alkyl iodides because a neutral ether is an even better leaving group than an iodide ion. |
| 18.61 |
The mechanism of Grignard addition to oxetane is the same as the mechanism of Grignard addition to epoxides, described in Section 18.5. The reaction proceeds at a reduced rate because oxetane is less reactive than ethylene oxide. The four-membered ring oxetane is less strained, and therefore more stable, than the three-membered ethylene oxide ring. |
| 18.62 |
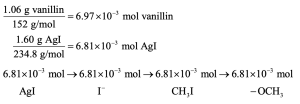
Thus, 6.97 × 10–3 mol of vanillin contain 6.81 x 10–3 mol of methoxyl groups. Since the ratio of moles vanillin to moles methoxyl is approximately 1:1, each vanillin contains one methoxyl group.
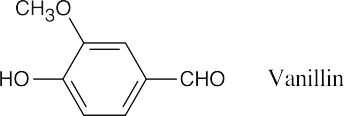
|
|
| 18.63 |
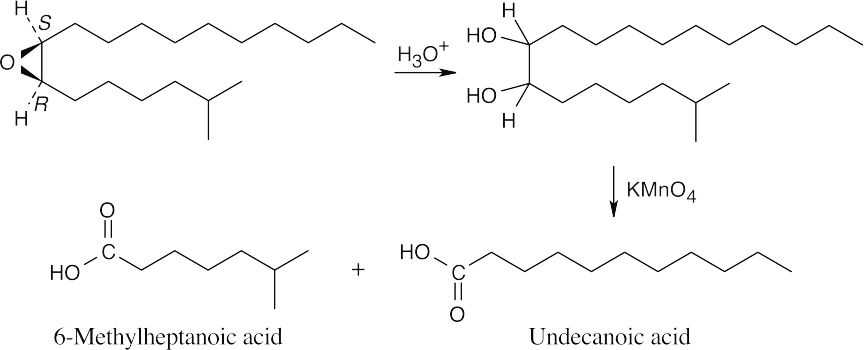
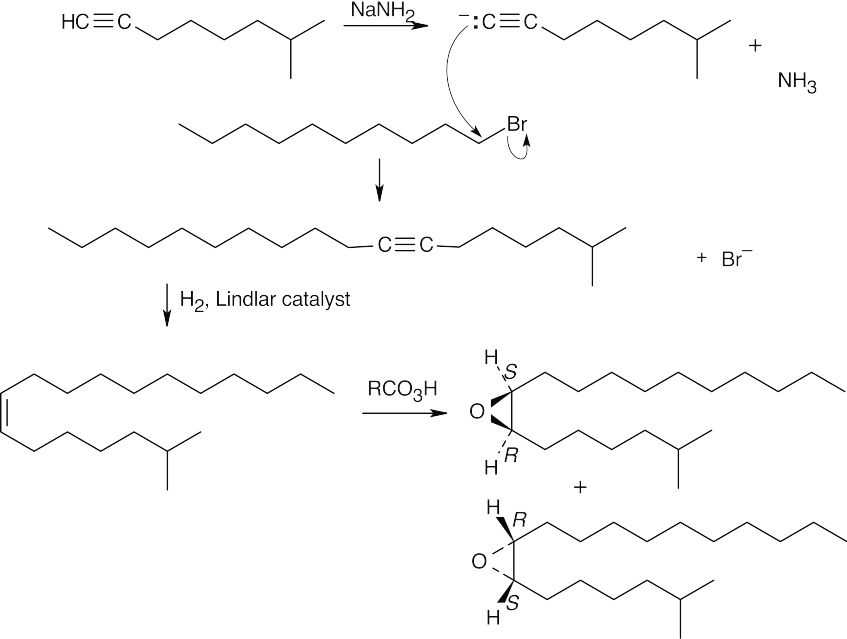
Disparlure, C19H38O, contains one degree of unsaturation, which the 1H NMR absorption at 2.8 δ identifies as an epoxide ring.
|
| 18.65 |
Use the aldehyde-forming reaction shown in the previous problem.
|
| 18.66 |
(a) CH3MgBr, ether;
(b) H2SO4, H2O;
(c) NaH, then CH3I;
(d) m-ClC6H4CO3H;
(e) –OH, H2O. |
| 18.67 |
| (a)
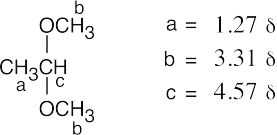
|
(b)
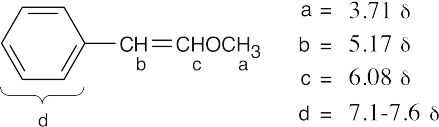
|
|
| 18.68 |
Step 1: Addition of hydride to the ketone.
Step 2: Displacement of bromide by the alkoxide anion.
The intermediate resulting from addition of H:– is similar to the intermediate in a Williamson ether synthesis. Intramolecular reaction occurs to form the epoxide. |
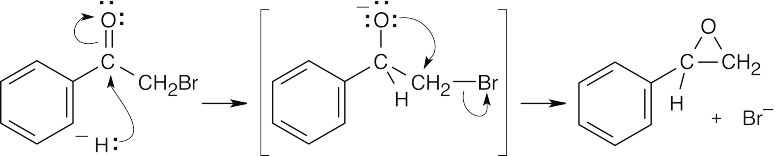
Preview of Carbonyl Chemistry Solutions to Problems
Solutions to Problems
| 18.69 |
According to the electrostatic potential maps, the carbonyl carbon of acetyl chloride is more electrophilic and the oxygen of acetone is more nucleophilic. This makes sense, because acetyl chloride has two electron-withdrawing groups that make its carbonyl carbon electron-poor and thus electrophilic. Because acetyl chloride has two electron-withdrawing groups, neither group is as nucleophilic as the carbonyl oxygen of acetone. |
| 18.70 |
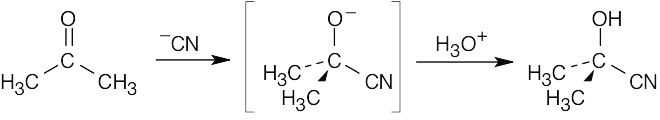
The reaction of cyanide ion with acetone is a nucleophilic addition reaction.
Cyanide anion adds to the positively polarized carbonyl carbon to form a tetrahedral intermediate. This intermediate is protonated to yield acetone cyanohydrin. |
| 18.71 |
| (a) |
This reaction is a nucleophilic acyl substitution. Ammonia adds to acetyl chloride, and chloride is eliminated, resulting in formation of an amide. |
| (b) |
In this nucleophilic addition reaction, addition of the nucleophile is followed by loss of water. |
| (c) |
Two molecules of cyclopentanone react in this carbonyl condensation. |
|
This file is copyright 2023, Rice University. All Rights Reserved.