
22 Chapter 22 – Carbonyl Alpha-Substitution Reactions Solutions to Problems
22.1–22.2Acidic hydrogens in the keto form of each of these compounds are bold. One of these hydrogens is removed by a base when an enolate is formed.
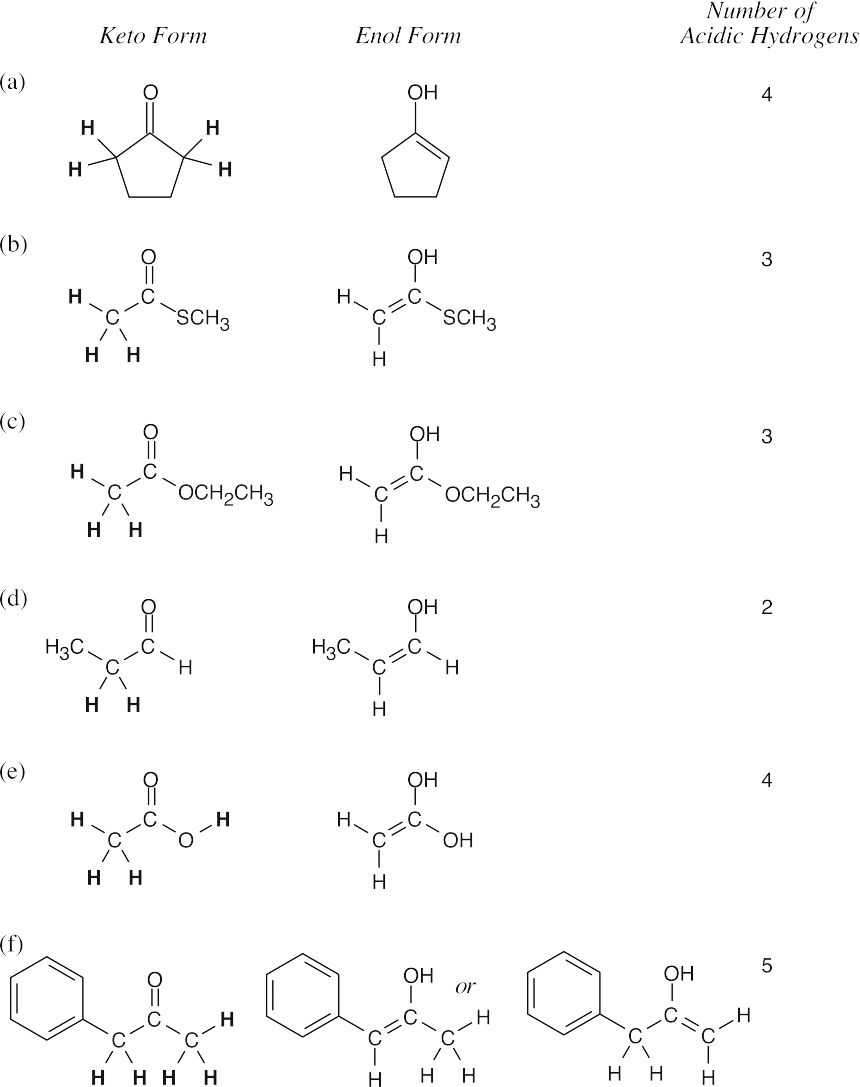
In (d) and (f), cis and trans enolates are possible.
22.3
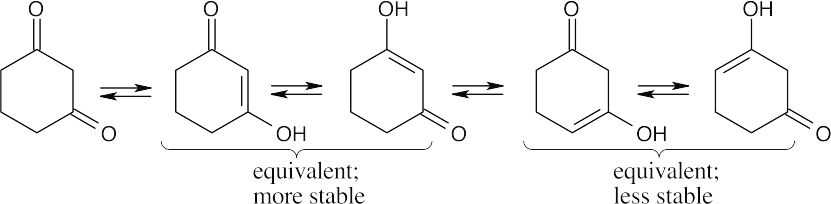
The first two monoenols are more stable because the enol double bond is conjugated with the carbonyl group.
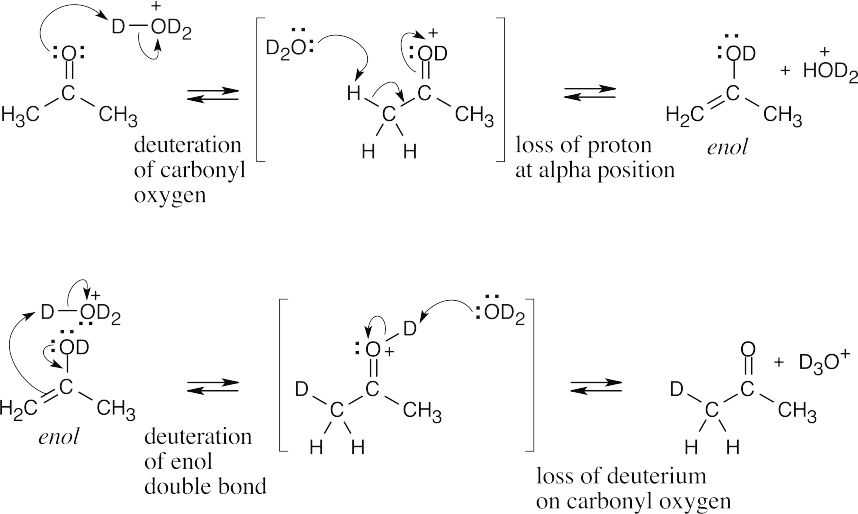
22.4
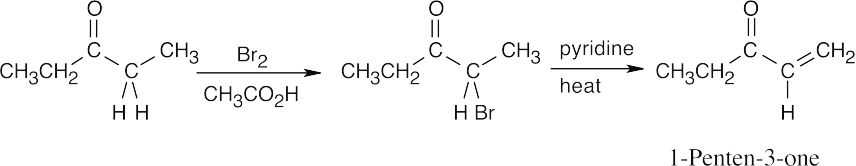
22.5Alpha-bromination, followed by dehydration using pyridine, yields the enone below.
22.6
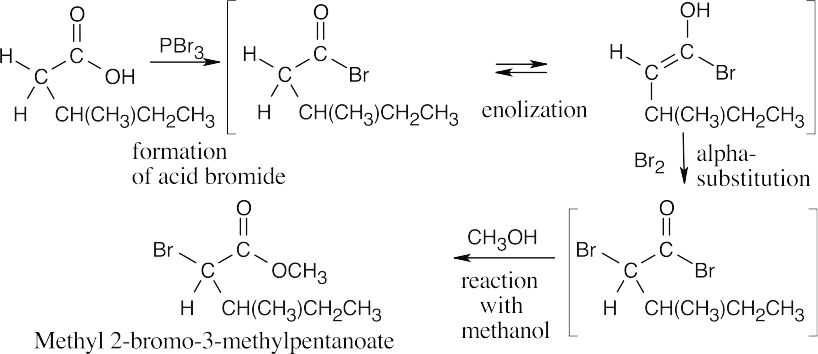
The mechanism of the ester-forming step is a nucleophilic acyl substitution, which was described in Chapter 21.
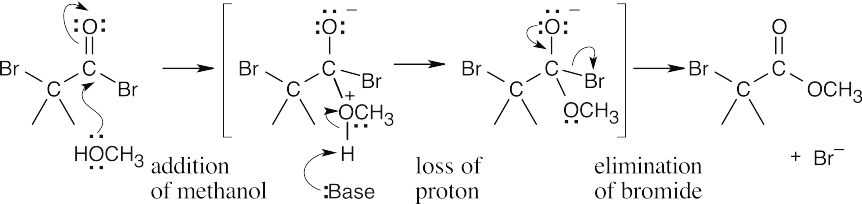
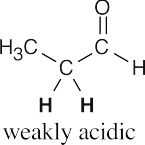
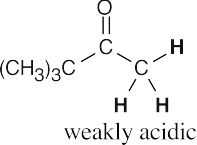
- Hydrogens α to one carbonyl group (or nitrile) are weakly acidic. Hydrogens α to two carbonyl groups are much more acidic, but they are not as acidic as carboxylic acid protons.
 (a)(b)(c)
(a)(b)(c)
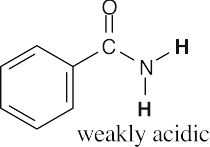
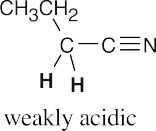
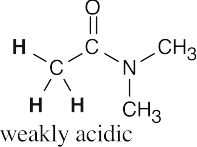 (d)(e)(f)
(d)(e)(f)
- Nitriles are weakly acidic because the nitrile anion can be stabilized by resonance involving the π bonds of the nitrile group.
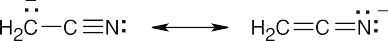
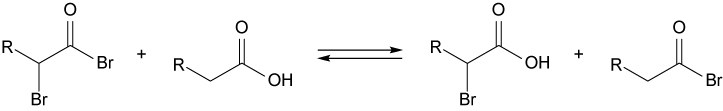
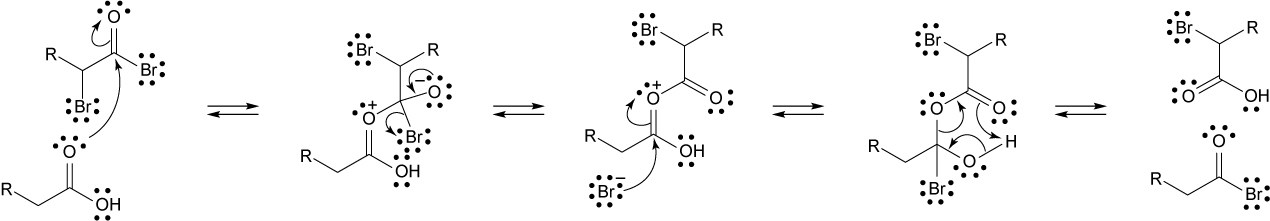
- Halogenation in an acid medium is acid-catalyzed because hydrogen ions are regenerated:
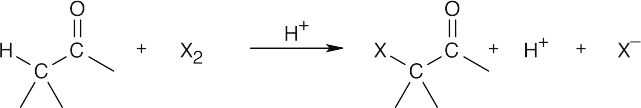
Halogenation in a basic medium is base-promoted because a stoichiometric amount of base is consumed:
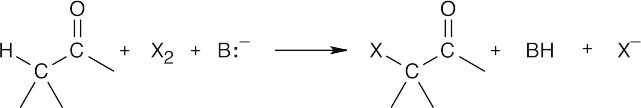
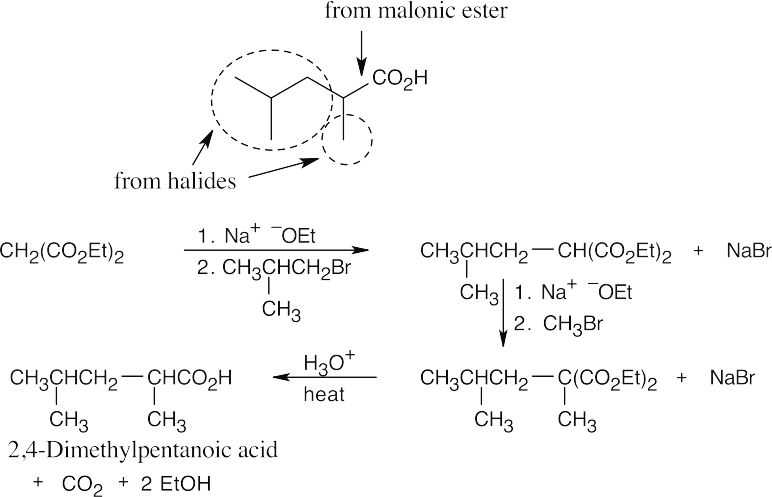
- The malonic ester synthesis converts a primary or secondary alkyl halide into a carboxylic acid with two more carbons (a substituted acetic acid). Identify the component that originates from malonic ester (the acid component). The rest of the molecule comes from the alkyl halide, which should be primary or methyl.
(a)
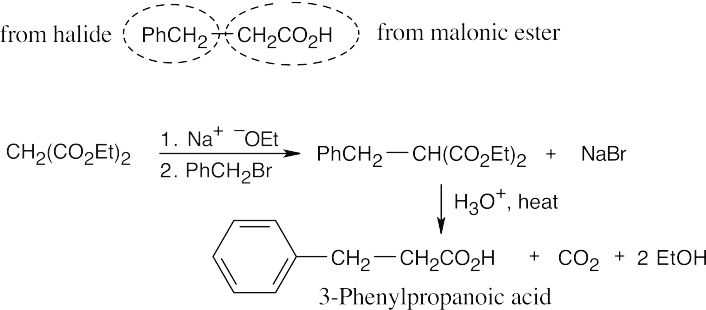
(b)
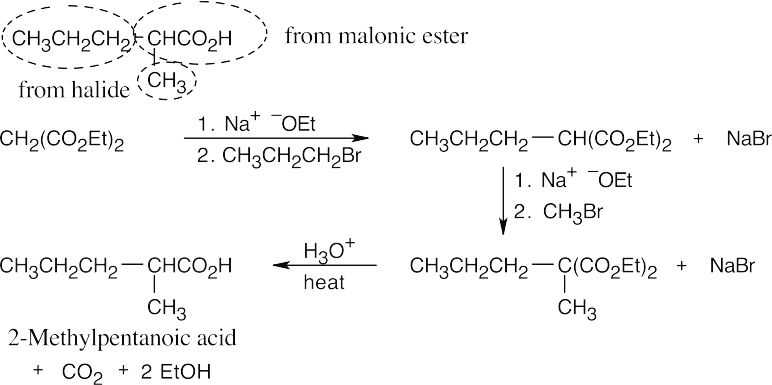
(c)
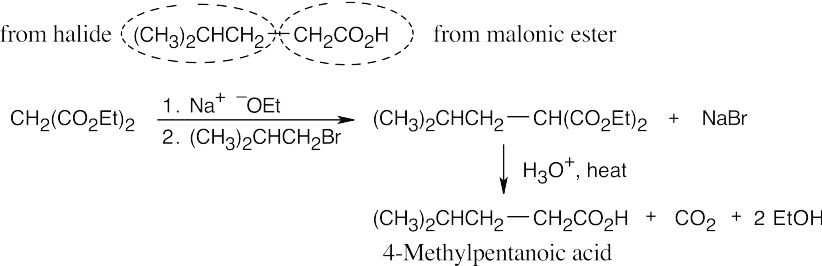
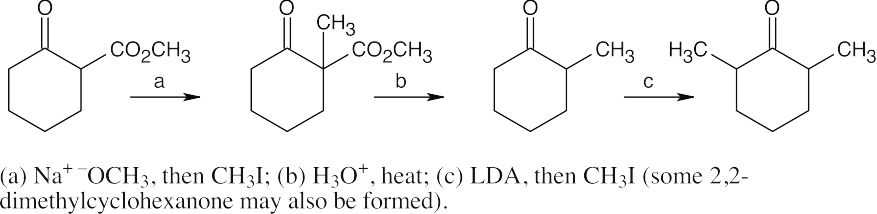
22.11 Since malonic ester has only two acidic hydrogen atoms, it can be alkylated only two times. Formation of trialkylated acetic acids is thus not possible.
22.12
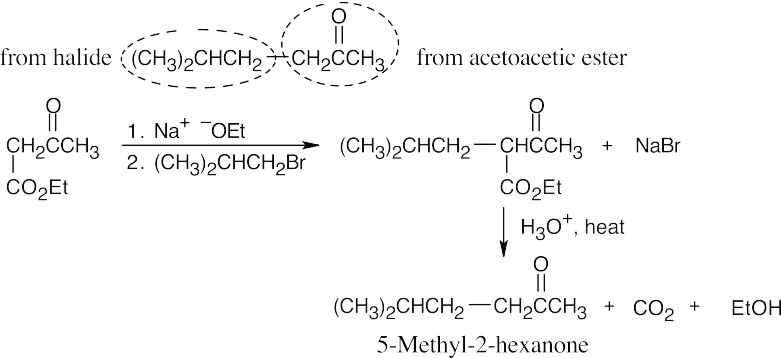
22.13 As in the malonic ester synthesis, you should identify the structural fragments of the target compound. The acetoacetic ester synthesis converts an alkyl halide to a methyl ketone (“substituted acetone”). The methyl ketone component comes from acetoacetic ester; the other component comes from a halide.
 (a)
(a)
(b)
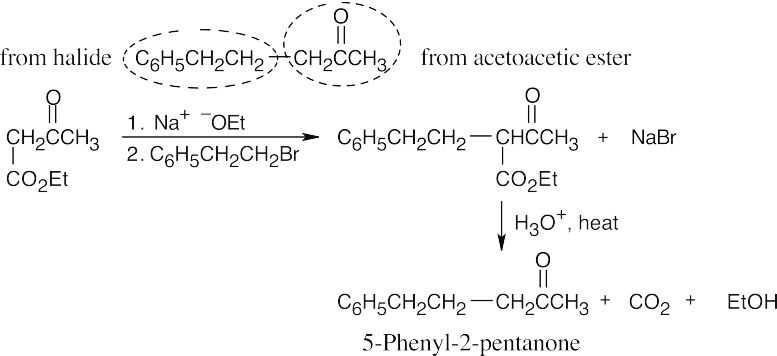
22.14 The acetoacetic ester synthesis can only be used for certain products:
- Three carbons must originate from acetoacetic ester. In other words, compounds of the type RCOCH3 can’t be synthesized by the reaction of RX with acetoacetic ester.
- Alkyl halides must be primary or methyl.
- The acetoacetic ester synthesis can’t be used to prepare compounds that are
trisubstituted at the α position.
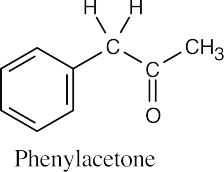
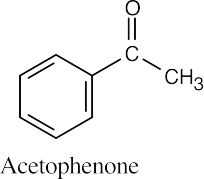
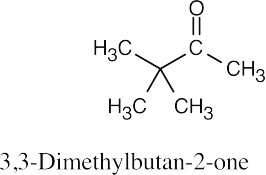 (a)(b)(c)
(a)(b)(c)
- Phenylacetone can’t be produced by an acetoacetic ester synthesis because bromobenzene, the necessary halide, does not enter into SN2 reactions. [See (2) above.]
- Acetophenone can’t be produced by an acetoacetic ester synthesis. [See (1) above.]
- 3,3-Dimethyl-2-butanone can’t be prepared because it is trisubstituted
at the α position.[See (3) above.]
22.15
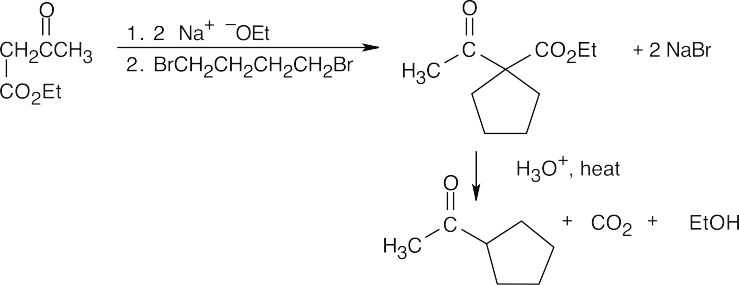
- Direct alkylation is used to introduce substituents α to an ester, ketone or nitrile. Look at the target molecule to identify these substituents. Alkylation is achieved by treating the starting material with LDA, followed by a primary halide.
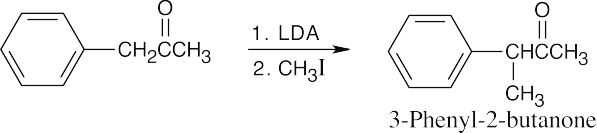 (a)
(a)
Alkylation occurs at the carbon next to the phenyl group because the phenyl group can help stabilize the enolate anion intermediate.
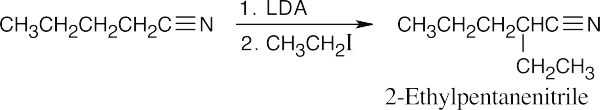 (b)
(b)
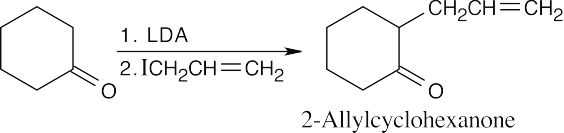 (c)
(c)
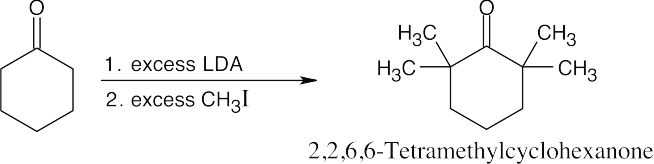
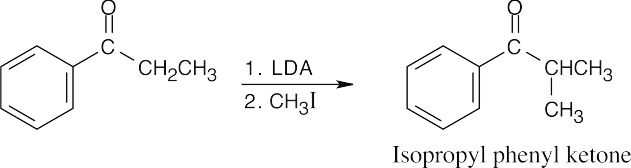
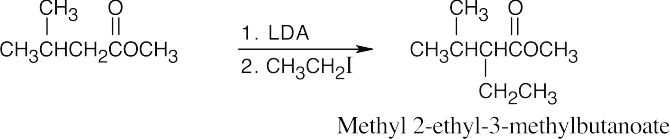 (f)
(f)
Additional Problems
Visualizing Chemistry
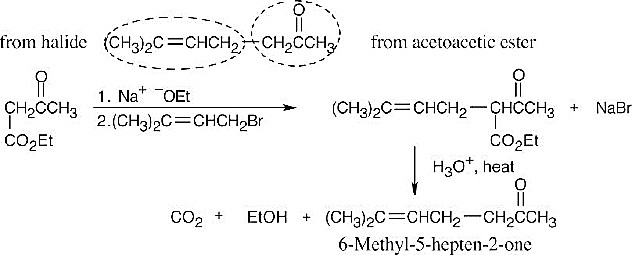 (a)Check to see if the target molecule is a methyl ketone or a substituted carboxylic acid. (The target molecule is a methyl ketone, and the reaction is an acetoacetic ester synthesis.) Next, identify the halide or halides that react with acetoacetic ester. (The halide is 1-bromo-3-methyl-2-butene.) Formulate the reaction, remembering to include a decarboxylation step.
(a)Check to see if the target molecule is a methyl ketone or a substituted carboxylic acid. (The target molecule is a methyl ketone, and the reaction is an acetoacetic ester synthesis.) Next, identify the halide or halides that react with acetoacetic ester. (The halide is 1-bromo-3-methyl-2-butene.) Formulate the reaction, remembering to include a decarboxylation step.
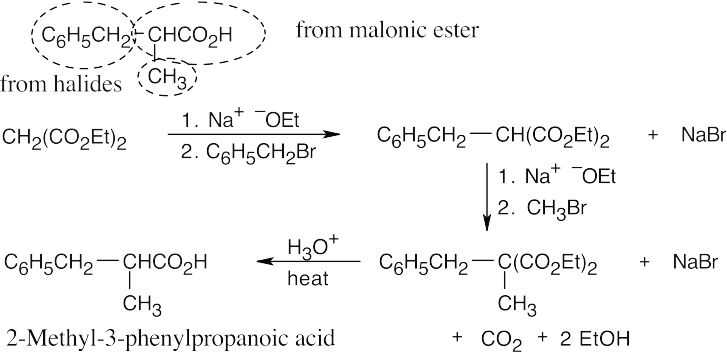
(b)This product is formed from the reaction of malonic ester with both benzyl bromide and bromomethane.
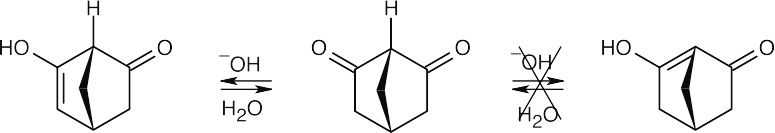
Ordinarily, β-diketones are acidic because they can form enolates that can be stabilized by delocalization over both carbonyl groups. In this case, loss of the proton at the bridgehead carbon doesn’t occur because the strained ring system doesn’t allow formation of the bridgehead double bond. Instead, enolization takes place in the opposite direction, and the diketone resembles acetone, rather than a β-diketone, in its pKa and degree of dissociation.
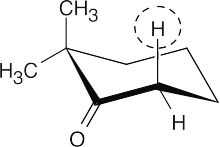
Enolization can occur on only one side of the carbonyl group because of the two methyl groups on the other side. The circled axial hydrogen is more acidic because the p orbital that remains after its removal is aligned for optimum overlap with the π electrons of the carbonyl oxygen.
Mechanism Problems
22.20 (a)
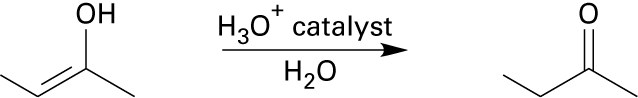
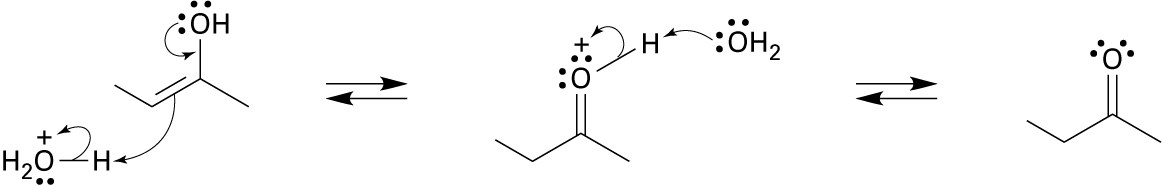
Mechanism:
(b)
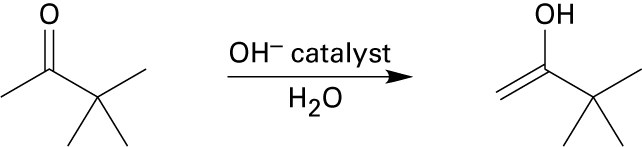
Mechanism:
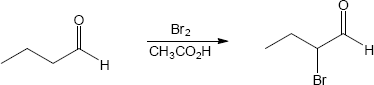 22.21 (a)
22.21 (a)
Mechanism:
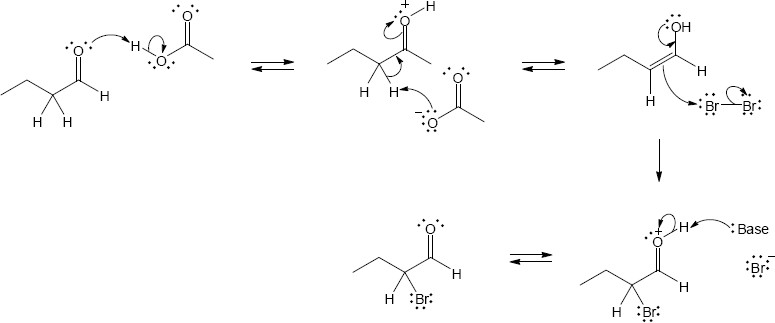
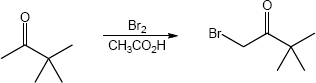 (b)
(b)
Mechanism:
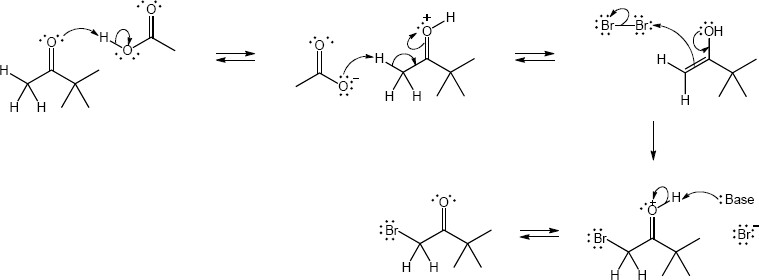
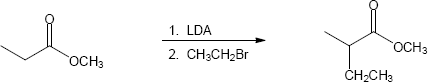 22.22 (a)
22.22 (a)
Mechanism:
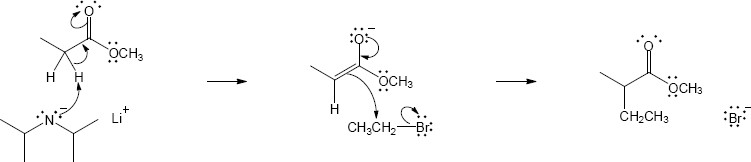
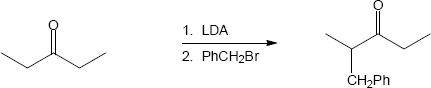 (b)
(b)
Mechanism:
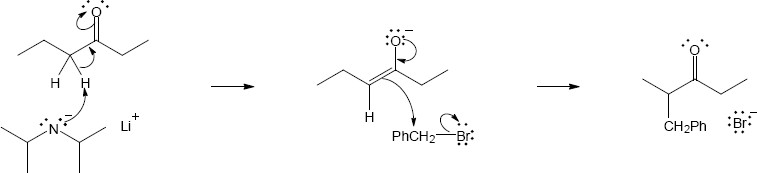
22.23(a)
 Mechanism:
Mechanism:
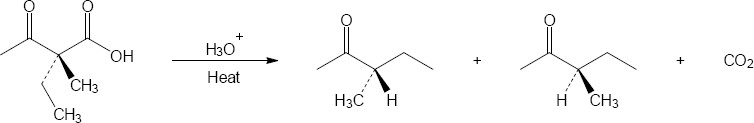
Note: The product mixture will not be optically active, but a racemic mixture. During the reaction, the chiral center becomes part of an achiral enol intermediate. When the enol tautomerizes, both enantiomeric ketones are formed in equal amounts.
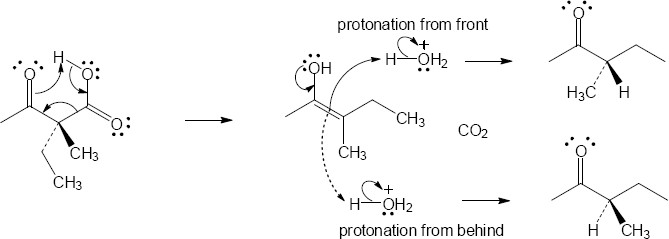
(b)
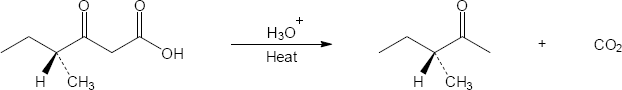
Mechanism:
Note: A single, optically active product is formed in this reaction. The chiral center in the starting material is never involved in the mechanism, so it remains unchanged.
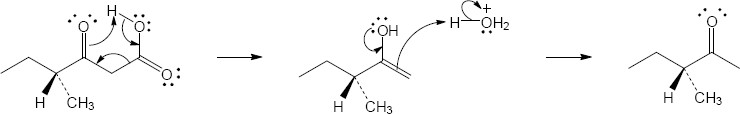
22.24
Mechanism:
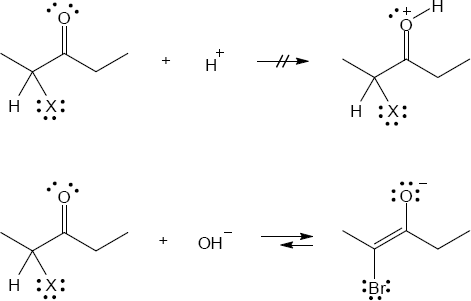 22.25 Note: Under acidic conditions, the ketone must form an enol that reacts with the halogen. The first step of enol formation is protonation of the carbonyl carbon. When there is a halogen α to the carbonyl, the inductive effect created by the halogen removes electron density from the carbonyl oxygen making it more difficult to protonate. This makes the enol more difficult to form and thus inhibits formation of an enol. When the reaction is performed under basic conditions, the inductive effect of the halogen removes electron density from the carbon to which it is attached, which in turn makes the proton attached to the same carbon more acidic. This facilitates formation of an enolate, making the addition of a second halogen faster than the first.
22.25 Note: Under acidic conditions, the ketone must form an enol that reacts with the halogen. The first step of enol formation is protonation of the carbonyl carbon. When there is a halogen α to the carbonyl, the inductive effect created by the halogen removes electron density from the carbonyl oxygen making it more difficult to protonate. This makes the enol more difficult to form and thus inhibits formation of an enol. When the reaction is performed under basic conditions, the inductive effect of the halogen removes electron density from the carbon to which it is attached, which in turn makes the proton attached to the same carbon more acidic. This facilitates formation of an enolate, making the addition of a second halogen faster than the first.
22.26
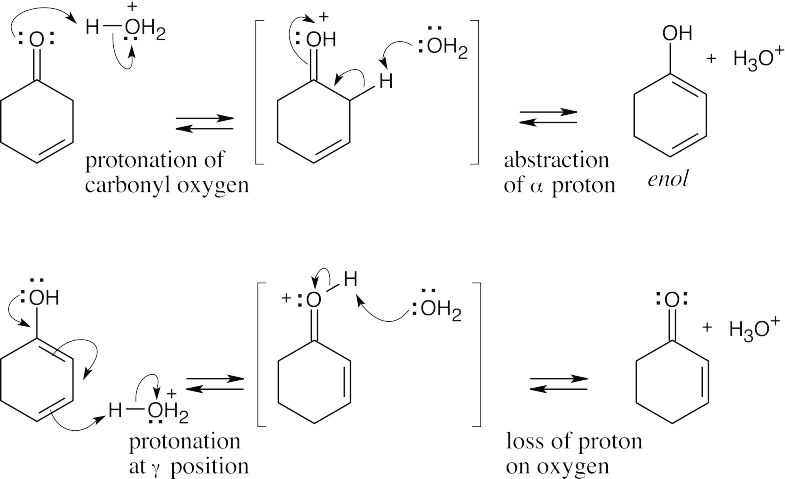
22.27
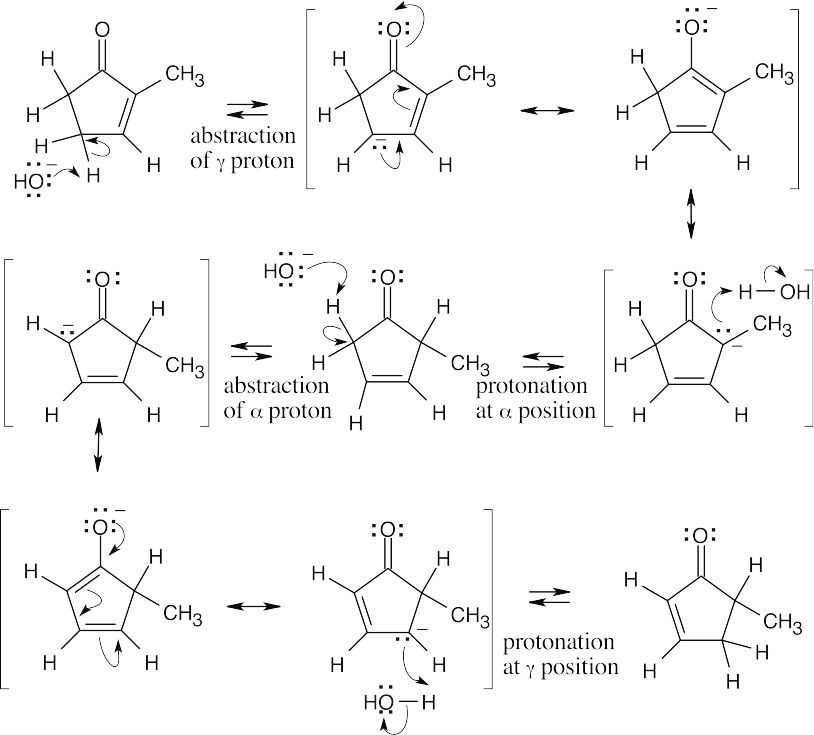
All protons in the five-membered ring can be exchanged by base treatment.
22.28
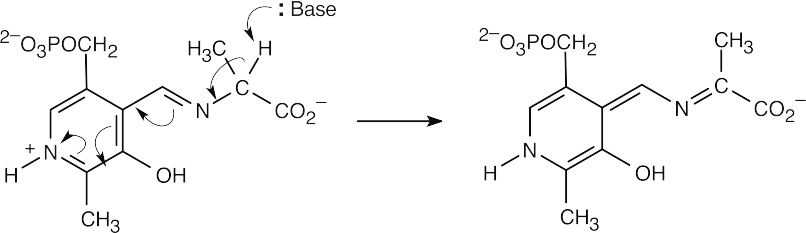
22.29 Decarboxylation, which takes place because of the stability of the resulting anion, is followed by protonation.
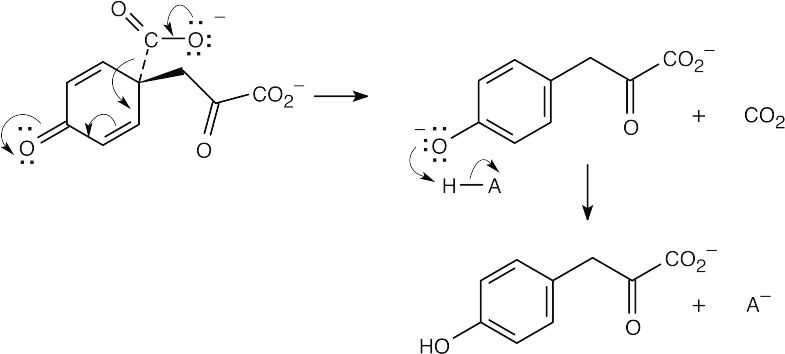
22.30
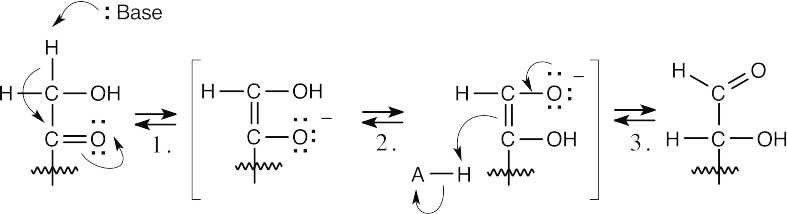
Step 1:Base-catalyzed enolization.
Step 2:Equilibration of two enolates by proton transfer.
Step 3:Protonation.
22.31
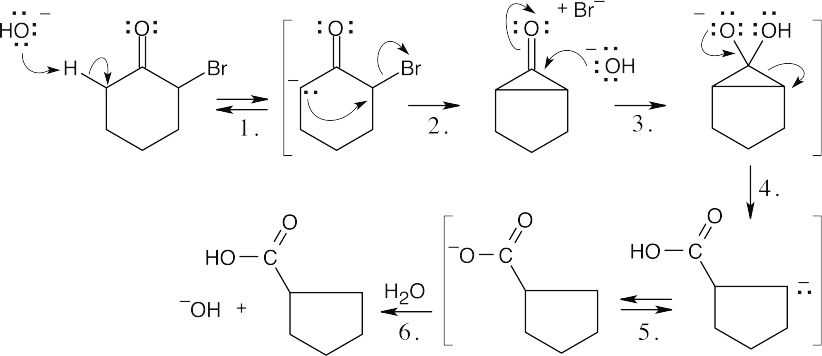
Step 1:Loss of proton at the α position.
Step 2:Displacement of bromide. Step 3:Nucleophilic addition of –OH. Step 4:Ring opening.
Step 5:Proton transfer.
Step 6:Protonation.
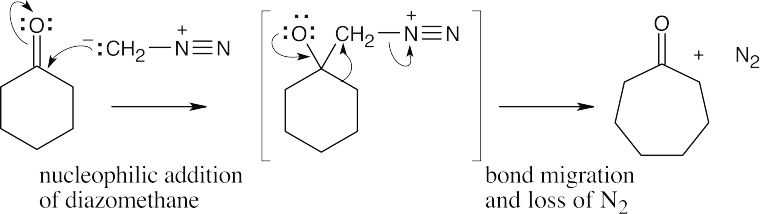
22.32
22.33 Laurene differs in stereochemical configuration from the observed product at the carbon α to the methylene group. Since this position is α to the carbonyl group in the precursor to laurene, enolization and isomerization must have occurred during the reaction. Isomerization of the ketone precursor is brought about by a reversible reaction with the basic Wittig reagent, which yields an equilibrium mixture of two diastereomeric ketones. One of the ketone isomers then reacts preferentially with the Wittig reagent to give only the observed product.
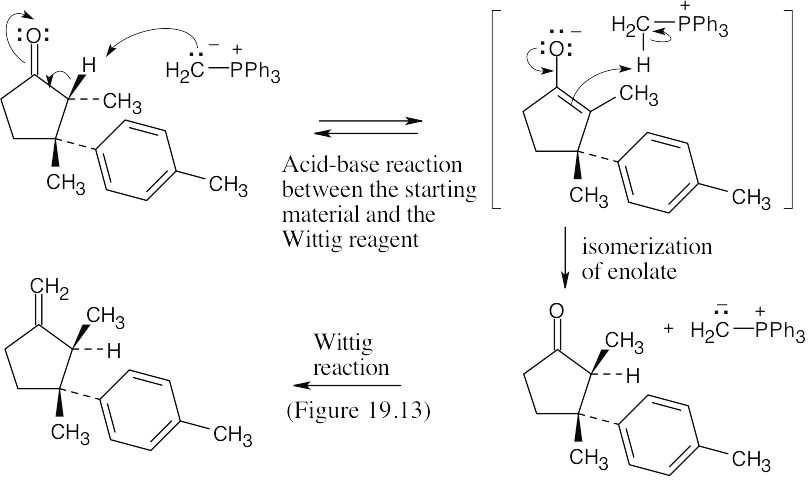
22.34
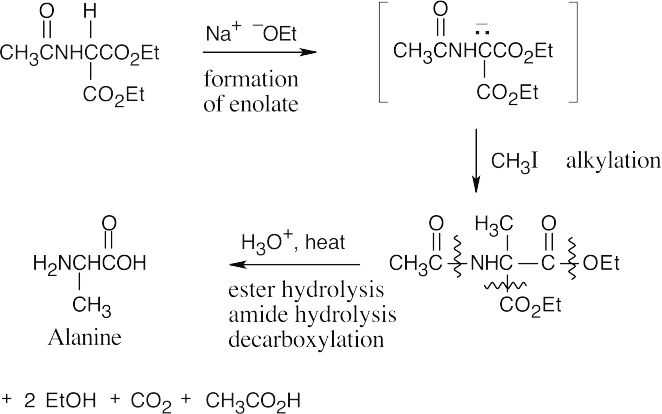
Acid hydrolyzes both ester bonds, as well as the amide bond, by mechanisms that were shown in Figure 21.9 and Section 21.6. Decarboxylation of the β-keto acid produces alanine.
22.35
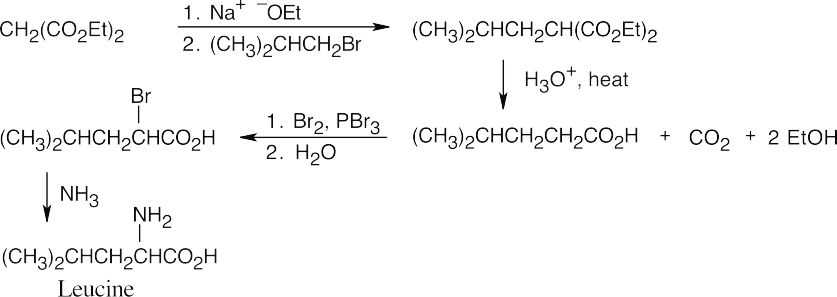
A malonic ester synthesis is used to form 4-methylpentanoic acid. Hell–Volhard– Zelinskii bromination of the acid, followed by reaction with ammonia, yields leucine. The last reaction is an SN2 displacement of bromide by ammonia.
22.36
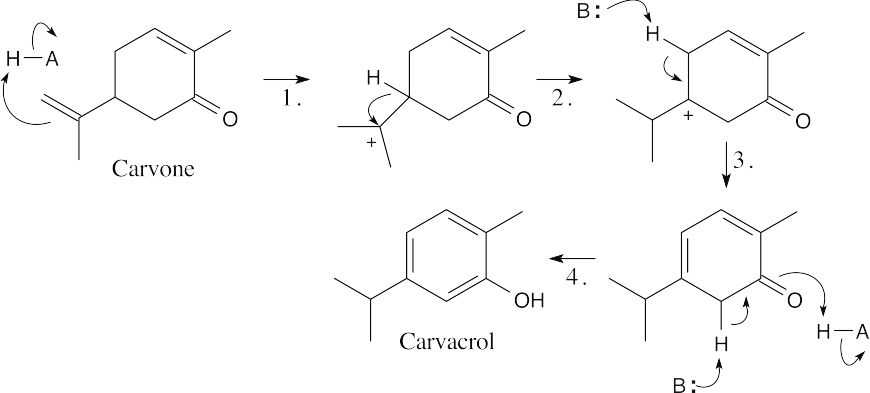
Step 1:Protonation. Step 2:Hydride shift. Step 3:Deprotonation.
Step 4:Enolization to form the aromatic ring.
Acidity of Carbonyl Compounds
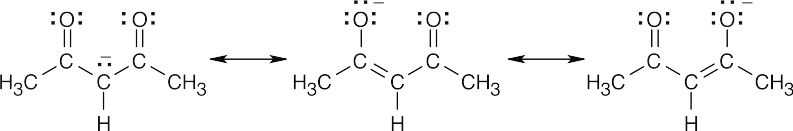
- Acidic hydrogens are bold. The most acidic hydrogens are the two between the carbonyl groups in (b) and the hydroxyl hydrogen in (c). The hydrogens in (c) that are bonded to the methyl group are acidic (draw resonance forms to prove it).
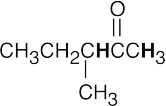
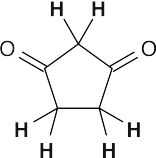
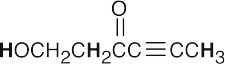 (a)(b)(c)
(a)(b)(c)
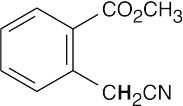
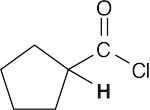
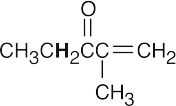 (d)(e)(f)
(d)(e)(f)
- Check your answer by using the pKas in Table 22.1.
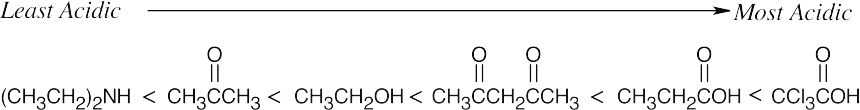
 (a)
(a)
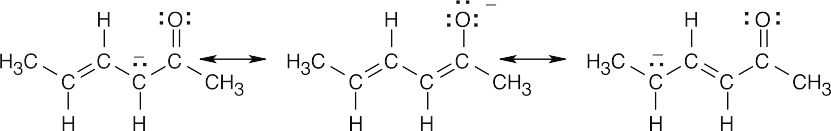 (b)
(b)
(c)
(d)
(e)
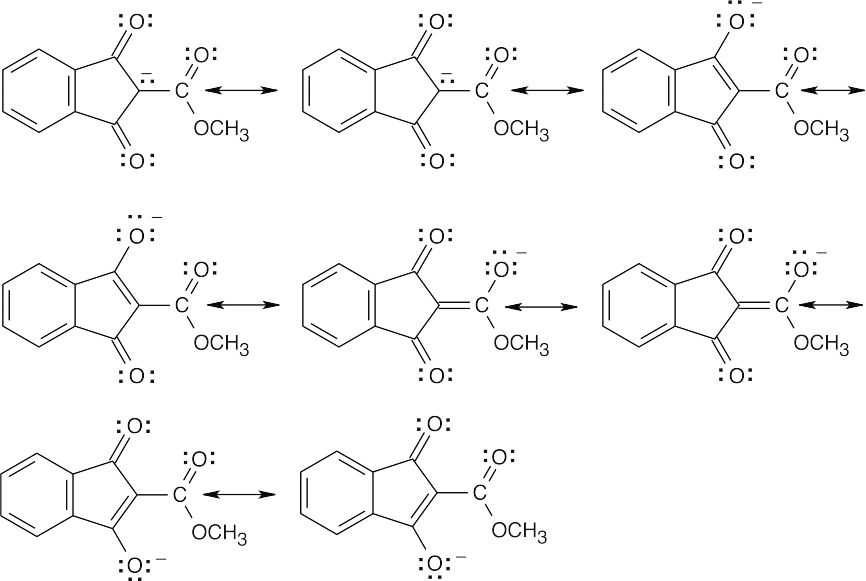
- Enolization at the γ position produces a conjugated enolate anion that is stabilized by delocalization of the negative charge over the π system of five atoms.
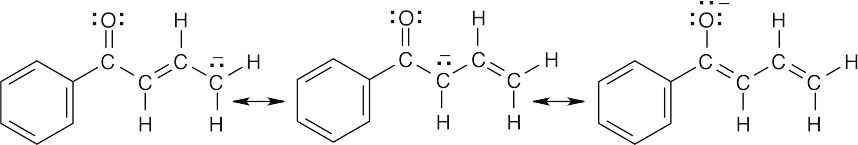
- The illustrated compound, 1-phenyl-2-propenone, doesn’t yield an anion when treated with a base because the hydrogen on the α carbon is vinylic and isn’t acidic (check Table 22.1 for acidity constants).
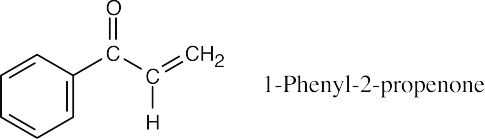
α-Substitution Reactions
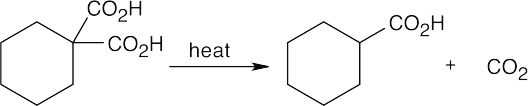 (a)
(a)
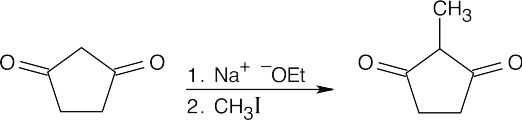 (b)
(b)
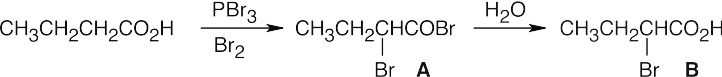 (c)
(c)
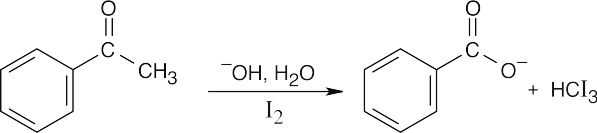 (d)
(d)
22.43
(a)
(b)
(c)
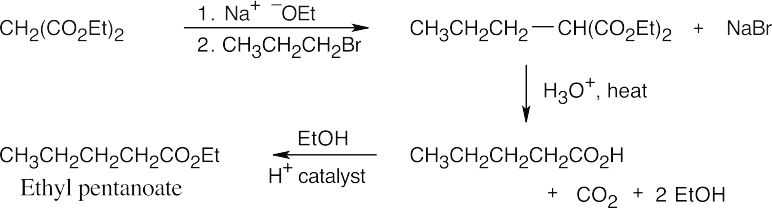
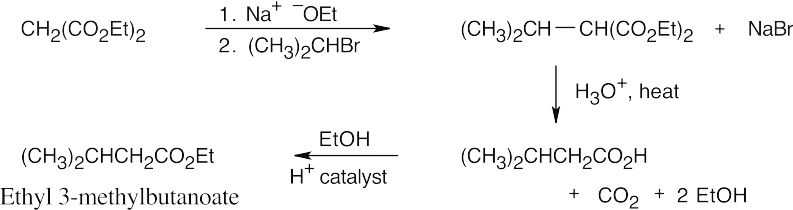
Some elimination product will also be formed.
(d)The malonic acid synthesis can’t be used to synthesize carboxylic acids that are trisubstituted at the alpha position.
- Look back to Problem 22.14, which describes compounds that can be prepared by an acetoacetic ester synthesis. Neither (a) or (c) are products of an acetoacetic ester synthesis because the halide component that would be needed for each synthesis doesn’t undergo SN2 reactions. Compound (b) can be prepared by the reaction of acetoacetic ester with
1,5-dibromopentane.
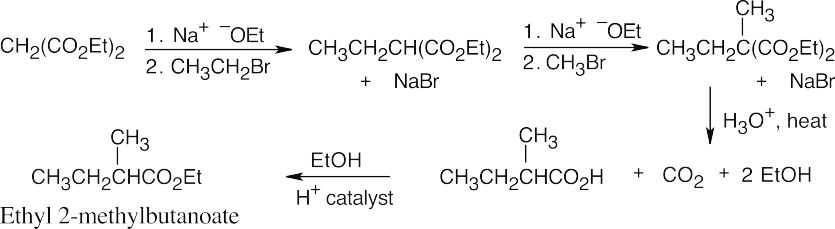
- Two alkylations are needed if the target molecule has two alkyl groups α to the carbonyl group.
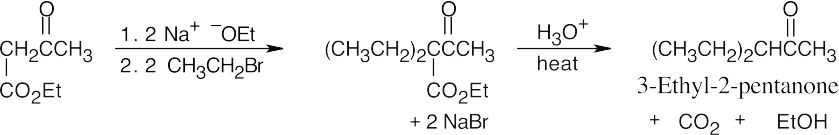 (a)
(a)
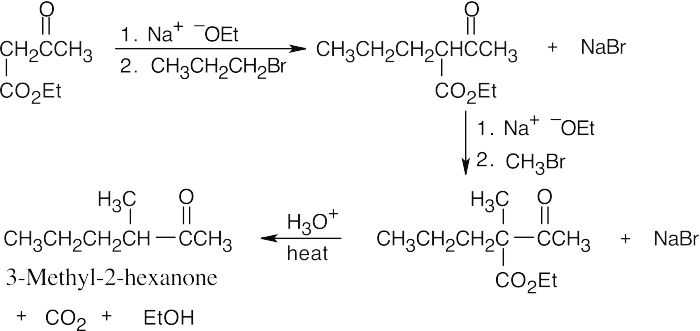 (b)
(b)
22.46 Use a malonic ester synthesis if the product you want is an α-substituted carboxylic acid or derivative. Use an acetoacetic acid synthesis if the product you want is an α– substituted methyl ketone.
 (a)
(a)
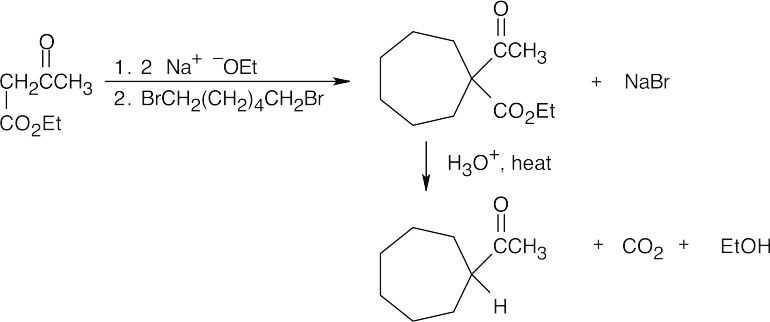 (b)
(b)
(c)
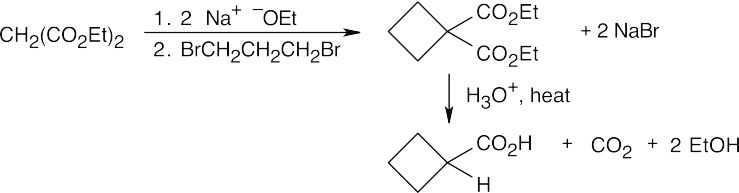
(d)
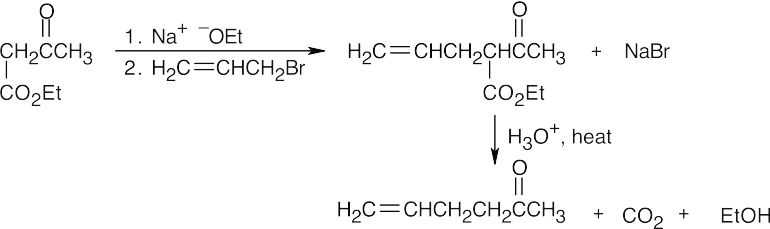
- The haloform reaction (Problem 22.42d) is an alpha-substitution reaction in which a methyl ketone is trihalogenated at the alpha position, and the trihalomethyl group is displaced by –OH. It is a test for methyl ketones.
Positive haloform reaction:Negative haloform reaction:
(a) 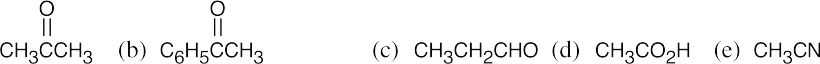
- First, treat geraniol with PBr3 to form (CH3)2C=CHCH2CH2C(CH3)=CHCH2Br (geranyl bromide).
(a)
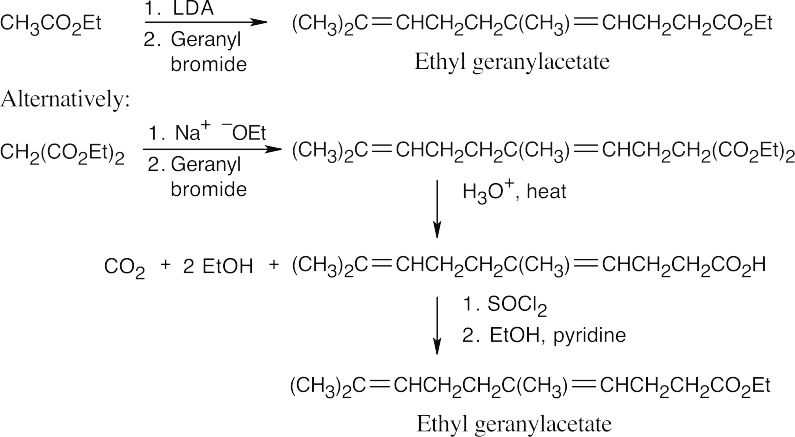
(b)
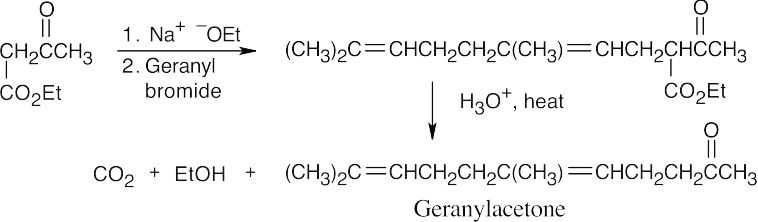
- Dialkylation of diethylmalonate:
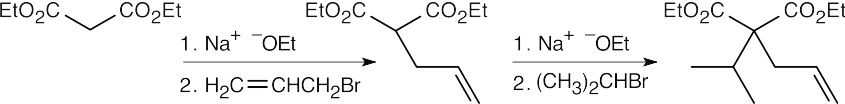
Nucleophilic acyl substitution:
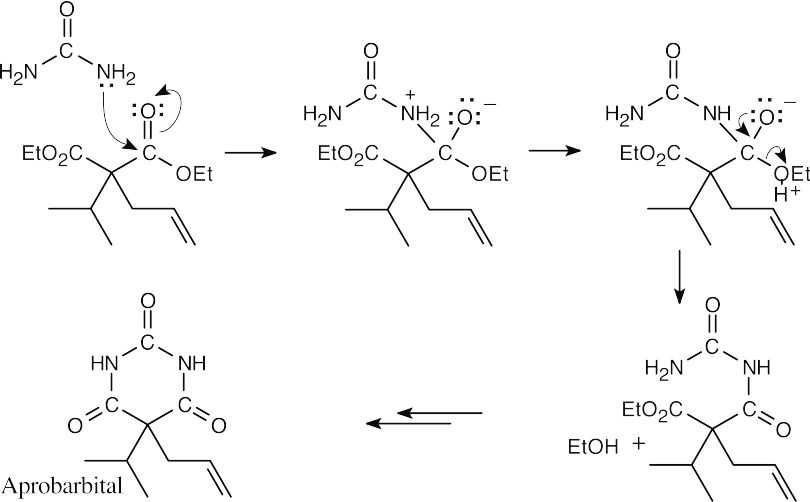
This series of steps is repeated to form the 6-membered ring.
General Problems
- When a compound containing acidic hydrogen atoms is treated with NaOD in D2O, all acidic hydrogens are gradually replaced by deuterium atoms. For each proton (atomic weight 1) lost, a deuteron (atomic weight 2) is added. Since the molecular weight of cyclohexanone increases by four after NaOD/D2O treatment (from 98 to 102), cyclohexanone contains four acidic hydrogen atoms.
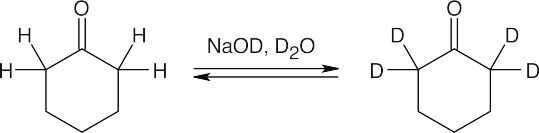
- Reaction of (R)-2-methylcyclohexanone with aqueous base is shown below. Reaction with aqueous acid proceeds by a related mechanism through an enol, rather than an enolate ion, intermediate.
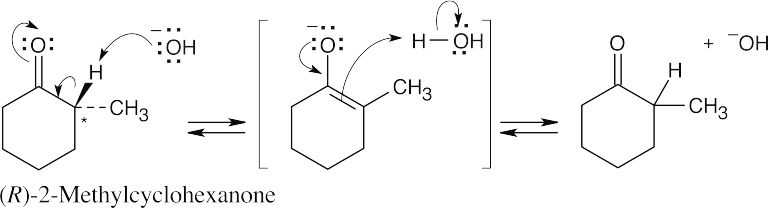
Carbon 2 loses its chirality when the enolate ion double bond is formed. Protonation occurs with equal probability from either side of the planar sp2-hybridized carbon 2, resulting in a racemic product.
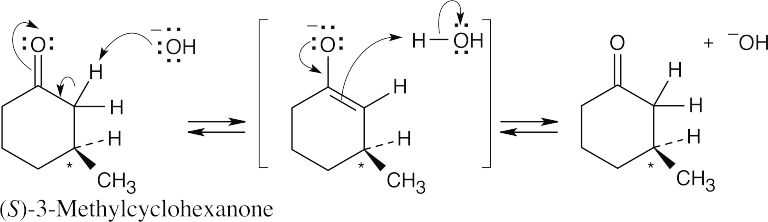
(S)-3-Methylcyclohexanone isn’t racemized by base because its chirality center is not involved in the enolization reaction.
- The Hell–Volhard–Zelinskii reaction involves formation of an intermediate acid bromide enol, with loss of stereochemical configuration at the chirality center. Bromination of (R)- 2-phenylpropanoic acid can occur from either face of the enol double bond, producing racemic 2-bromo-2-phenylpropanoic acid. If the molecule had a chirality center that didn’t take part in enolization (Problem 22.35), the product would be optically active.
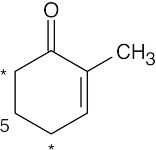
- Protons α to a carbonyl group or γ to an enone carbonyl group are acidic (Problem 22.39). Thus for 2-methyl-2-cyclopentenone, protons at the starred positions are acidic.
Isomerization of a 2-substituted 2-cyclohexenone to a 6-substituted 2-cyclohexenone requires removal of a proton from the 5-position of the 2-substituted isomer. Since protons in this position are not acidic, double bond isomerization does not occur.
(a)
(b)
(c)
(d)
(e)
(f)
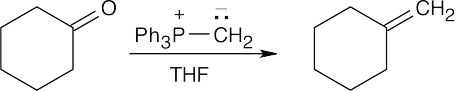
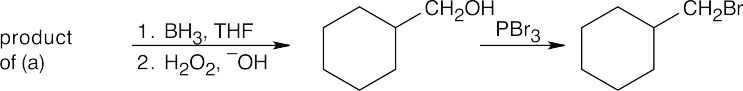
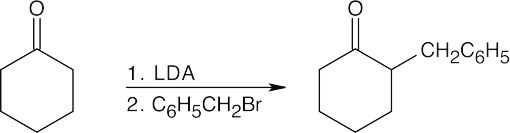
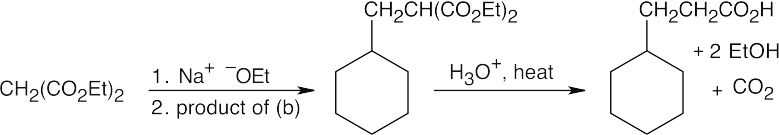
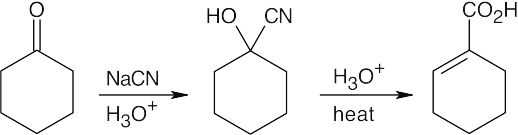
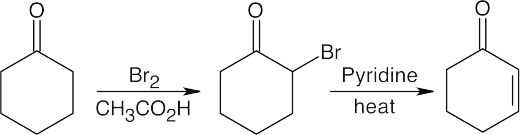 Warm aqueous acid both hydrolyzes the nitrile and dehydrates the alcohol.
Warm aqueous acid both hydrolyzes the nitrile and dehydrates the alcohol.
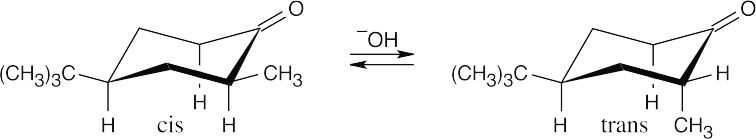
- Treatment of either the cis or trans isomer with base causes enolization α to the carbonyl group and results in loss of configuration at the α position. Reprotonation at carbon 2 produces either of the diastereomeric 4-tert-butyl-2-methylcyclohexanones. In both diastereomers the tert-butyl group of carbon 4 occupies the equatorial position for steric reasons. The methyl group of the cis isomer is also equatorial, but the methyl group of the trans isomer is axial. The trans isomer is less stable because of 1,3-diaxial interactions of the methyl group with the ring hydrogens.
- (a)Reaction with Br2 at the α position occurs only with aldehydes and ketones, not with esters.
- Aryl halides can’t be used in malonic ester syntheses because they don’t undergo SN2 reactions.
- The product of this reaction sequence, H2C=CHCH2CH2COCH3, is a methyl ketone, not a carboxylic acid.
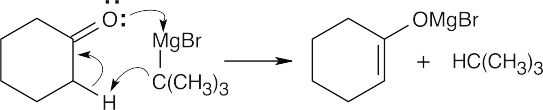
- The reaction of cyclohexanone and tert-butylmagnesium bromide gives the expected carbonyl addition product. The yield of the tert-butylmagnesium bromide addition product is very low, however, because of the difficulty of approach of the bulky tert- butyl Grignard reagent to the carbonyl carbon. More favorable is the acid-base reaction between the Grignard reagent and the acidic carbonyl α proton.
When D3O+ is added to the reaction mixture, the deuterated ketone is produced.
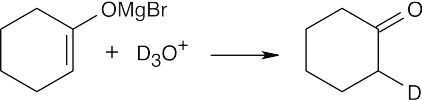
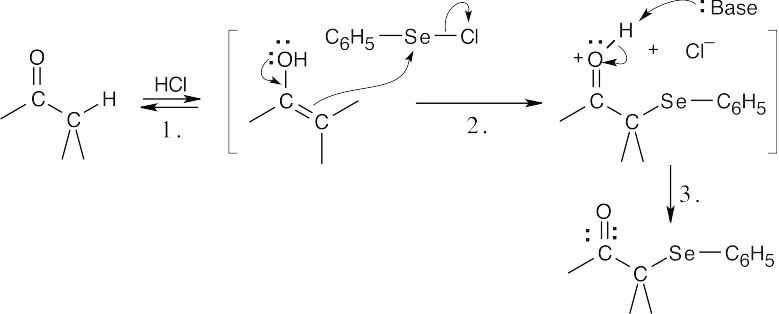
Step 1:Acid-catalyzed enolization (Figure 22.2).
Step 2:Attack of enol π electrons on phenylselenyl chloride, with loss of Cl–.
Step 3:Loss of proton.
- Start at the end of the sequence of reactions and work backwards.
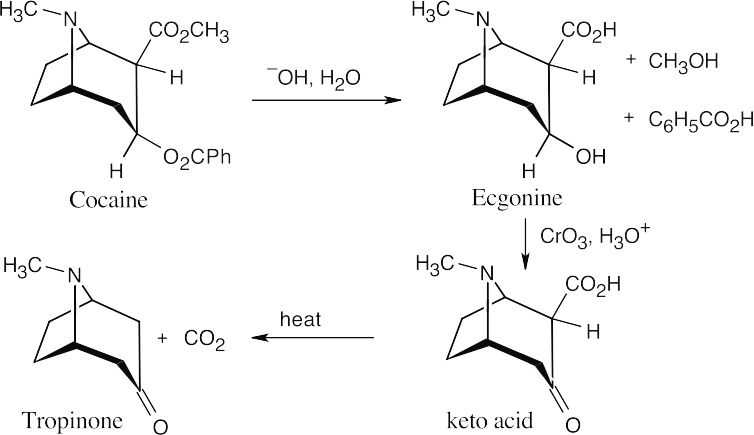
- Because the keto acid C9H13NO3 loses CO2 on heating, it must be a β-keto acid. Neglecting stereoisomerism, we can draw the structure of the β-keto acid as:
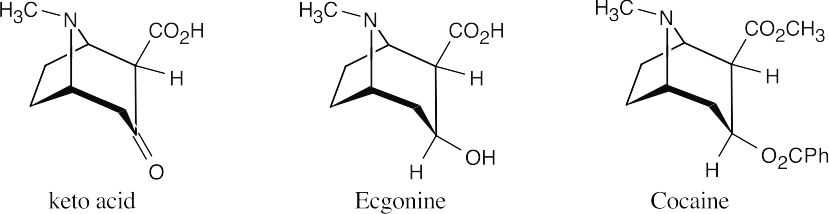
- When ecgonine (C9H15NO3) is treated with CrO3, the keto acid C9H13NO3 is produced. Since CrO3 is used for oxidizing alcohols to carbonyl compounds, ecgonine has the structure shown above. Again, the stereochemistry is unspecified.
- Ecgonine contains carboxylic acid and alcohol functional groups. The other products of hydroxide treatment of cocaine are a carboxylic acid (benzoic acid) and an alcohol (methanol). Cocaine thus contains two ester functional groups, which are saponified on reaction with hydroxide.
The complete reaction sequence:
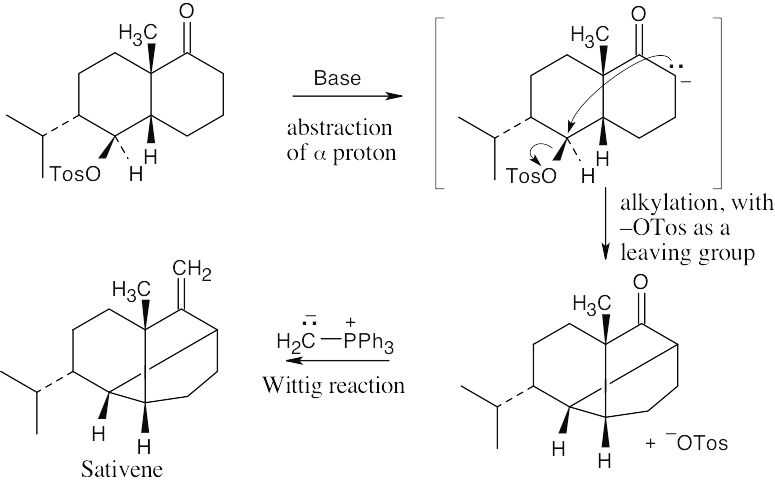
- The key step is an intramolecular alkylation reaction of the ketone α-carbon, with the tosylate in the second ring serving as the leaving group.
- This sequence resembles the one shown in Problem 22.49.
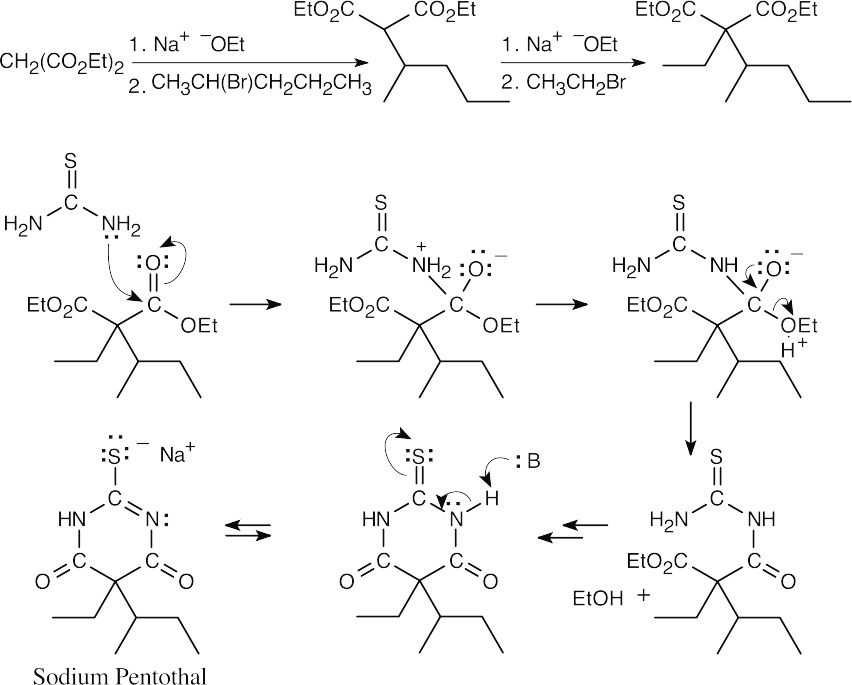
The series of steps is repeated to form the 6-membered ring.
This file is copyright 2023, Rice University. All Rights Reserved.
