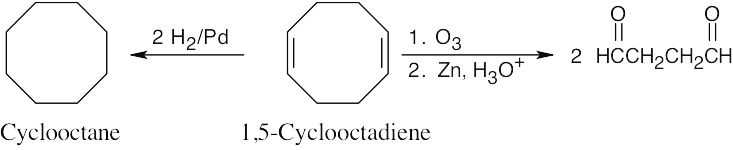
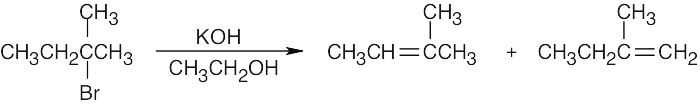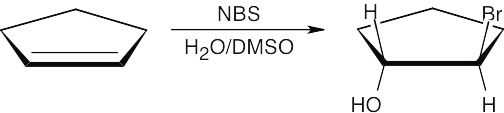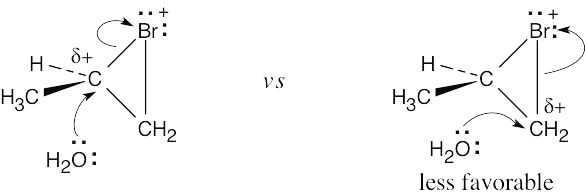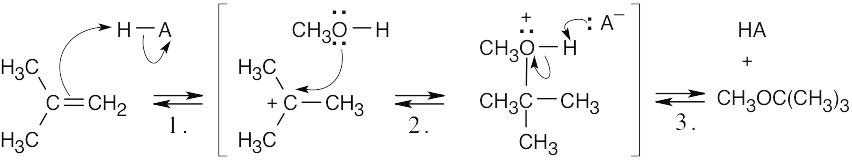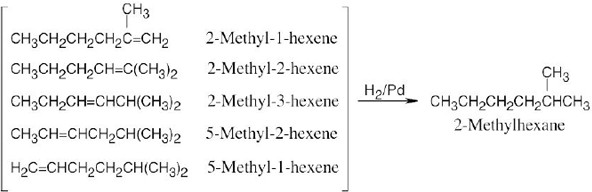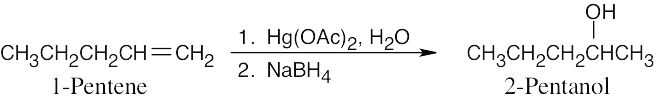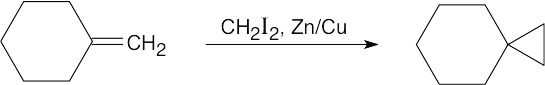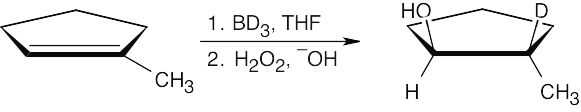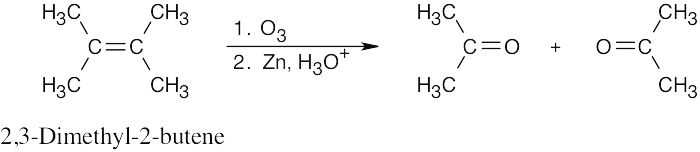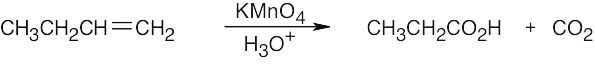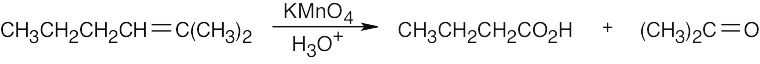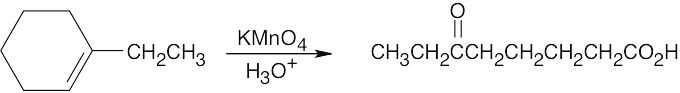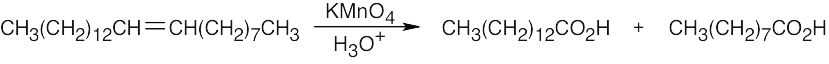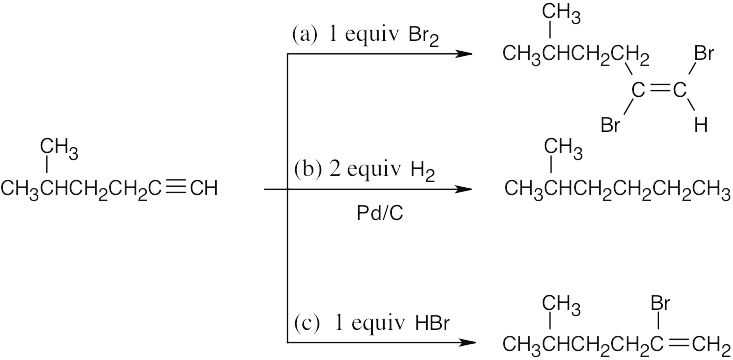8 Chapter 8 – Alkenes: Reactions and Synthesis Solutions to Problems
Chapter 8 – Alkenes: Reactions and Synthesis
Solutions to Problems
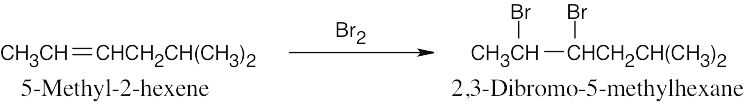
| 8.1 |
Dehydrobromination may occur in either of two directions to yield a mixture of products. |
| 8.2 |
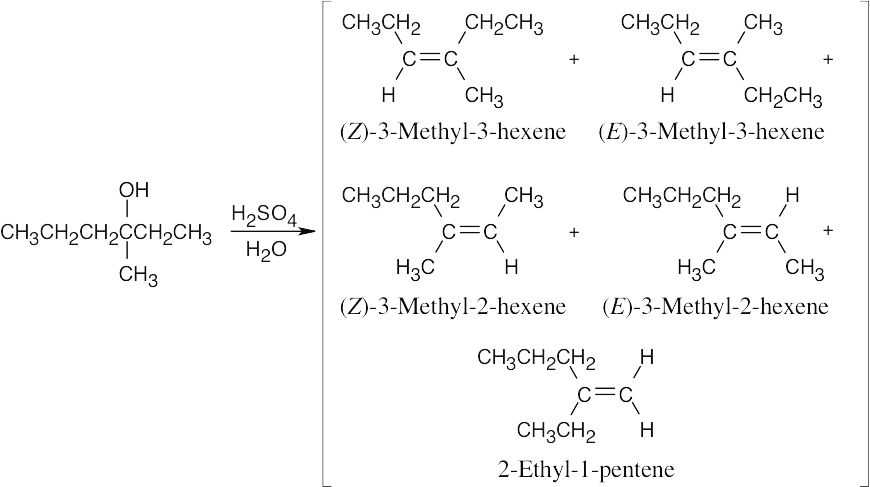
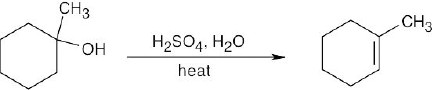
Five alkene products, including E, Z isomers, might be obtained by dehydration of 3- methyl-3-hexanol. |
| 8.3 |
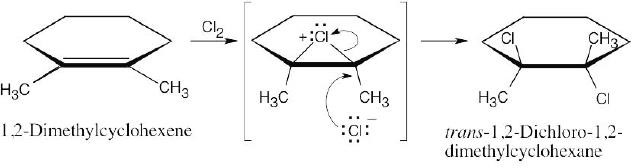
The chlorines are trans to one another in the product, as are the methyl groups. |
| 8.4 |
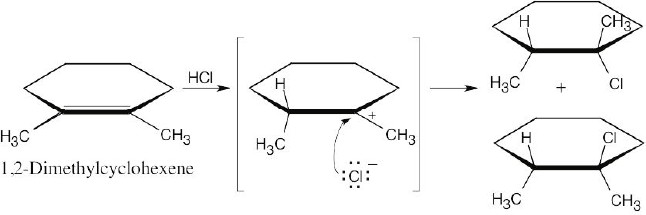
Addition of hydrogen halides involves formation of an open carbocation, not a cyclic halonium ion intermediate. The carbocation, which is sp2-hybridized and planar, can be attacked by chloride from either top or bottom, yielding products in which the two methyl groups can be either cis or trans to each other. |
| 8.5 |
–Br and –OH are trans in the product. |
| 8.6 |
Reaction of the alkene with Br2 (formed from NBS) produces a cyclic bromonium ion. When this bromonium ion is opened by water, a partial positive charge develops at the carbon whose bond to bromine is being cleaved.
Since a secondary carbon can stabilize this charge better than a primary carbon, opening of the bromonium ion occurs at the secondary carbon to yield the Markovnikov product. |
| 8.7 |
Keep in mind that oxymercuration is equivalent to Markovnikov addition of H2O to an alkene.
| (a)
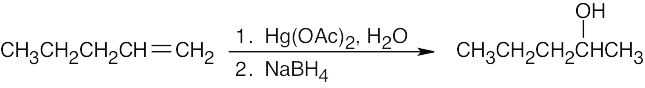
|
| (b)
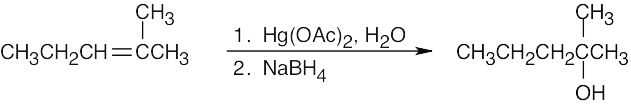
|
|
| 8.8 |
Think backwards to select the possible alkene starting materials for the alcohols pictured.
| (a)
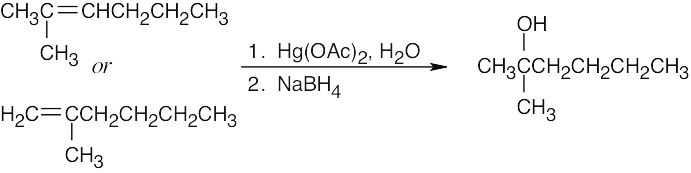
|
| (b)
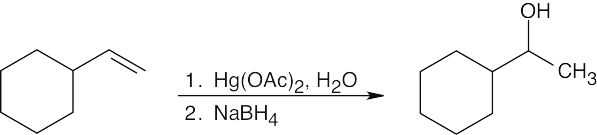
|
Oxymercuration occurs with Markovnikov orientation. |
| 8.9 |
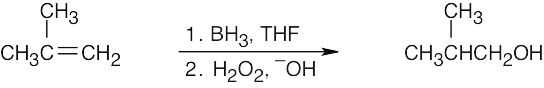
Hydroboration/oxidation occurs with non-Markovnikov regiochemistry to give products in which –OH is bonded to the less highly substituted carbon.
| (a)
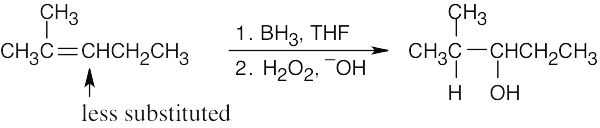
|
| (b)
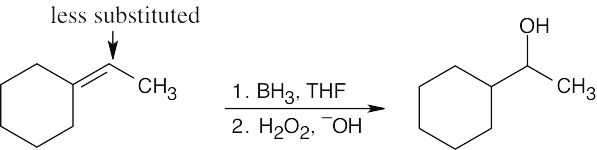
|
|
| 8.10 |
As described in Worked Example 8.2, the strategy in this sort of problem begins with a look backward. In more complicated syntheses this approach is essential, but even in problems in which the functional group(s) in the starting material and the reagents are known, this approach is effective.
All the products in this problem result from hydroboration/oxidation of a double bond. The –OH group is bonded to the less substituted carbon of the double bond in the starting material.
| (a)
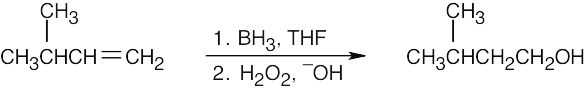
|
| (b)
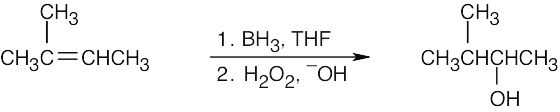
This product can also result from oxymercuration of the starting material in (a). |
| (c)
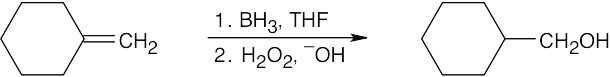
|
|
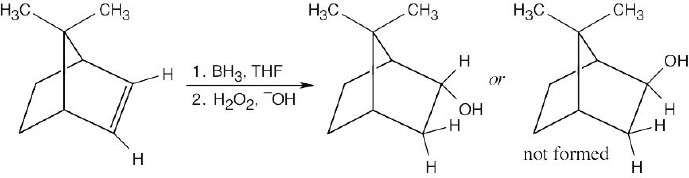
| 8.11 |
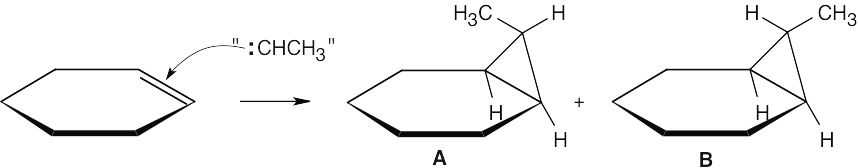
The drawings below show the transition states resulting from addition of BH3 to the double bond of the cycloalkene. Addition can occur on either side of the double bond.
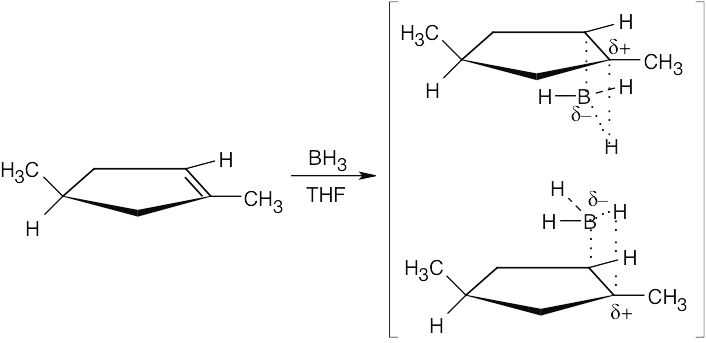
Reaction of the two neutral alkylborane adducts with hydrogen peroxide gives two alcohol isomers. In one isomer, the two methyl groups have a cis relationship, and in the other isomer they have a trans relationship.
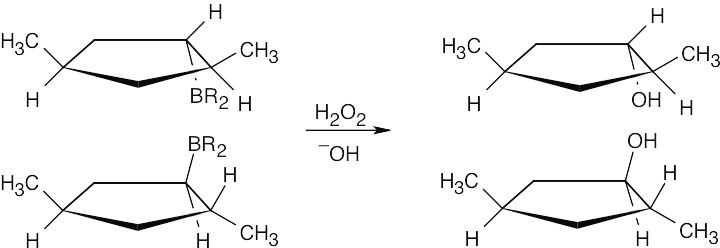
|
|
| 8.12 |
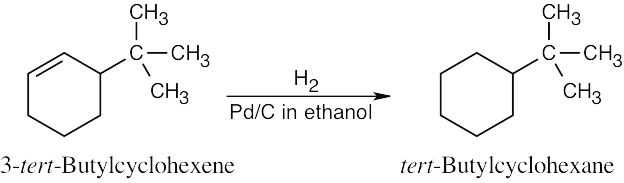
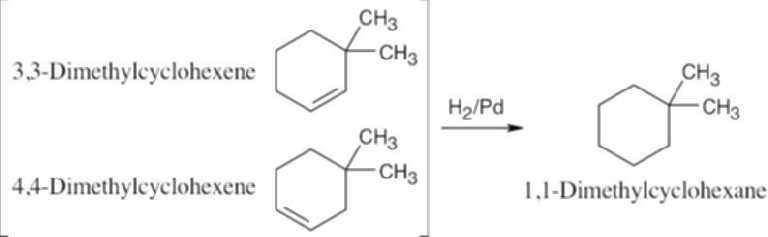
Catalytic hydrogenation produces alkanes from alkenes.
|
| 8.13 |
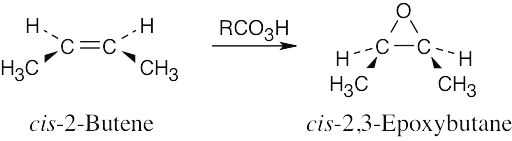
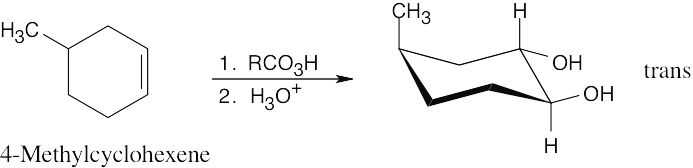
Epoxidation using m-chloroperoxybenzoic acid (RCO3H) is a syn addition of oxygen to a double bond. The original bond stereochemistry is retained.
In the epoxide product, as in the alkene starting material, the methyl groups are cis. |
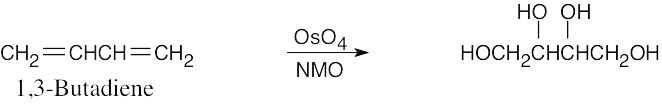
| 8.14 |
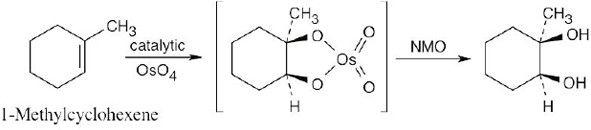
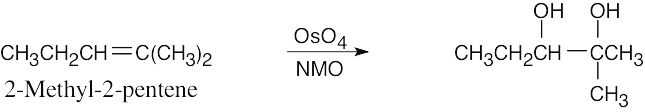
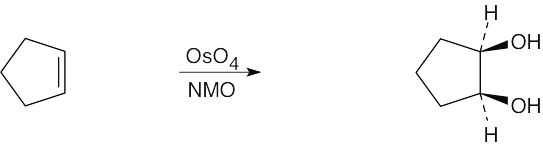
Reaction of an alkene with a catalytic amount of OsO4, in the presence of N-morpholine N-oxide (NMO), yields a diol product. To pick a starting material for these products, choose an alkene that has a double bond between the diol carbons. The products in (b) and (c) can also be formed by ring opening of an epoxide formed either from a peroxyacid or from a halohydrin.
|
| 8.15 |
Both sets of reactants cleave double bonds. Aqueous KMnO4 produces a carboxylic acid from a double bond carbon that is monosubstituted and a ketone from a double bond carbon that is disubstituted. Ozone produces an aldehyde from a double bond carbon that is monosubstituted and a ketone from a double bond carbon that is disubstituted. If the double bond is part of a ring, both carbonyl groups occur in the same product molecule.
| (a)
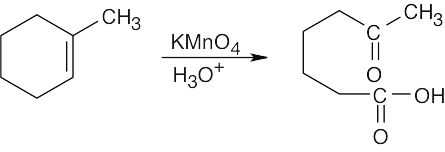
|
| (b)
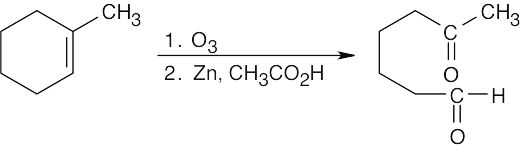
|
|
| 8.16 |
Orient the fragments so that the oxygens point toward each other. Remove the oxygens, and draw a double bond between the remaining carbons.
| (a)

|
| (b)
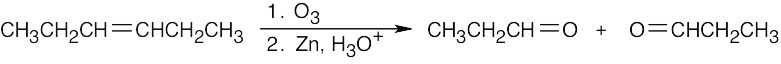
|
|
| 8.17 |
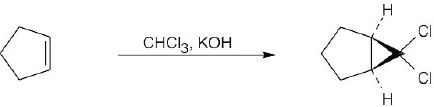
Reaction of a double bond with chloroform under basic conditions gives a product with a cyclopropane ring in which one of the carbons has two chlorine atoms bonded to it. Reaction of a double bond with CH2I2 yields a product with a cyclopropane ring that has a –CH2– group.
| (a)
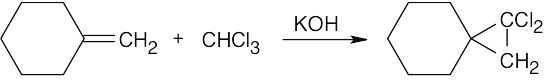
|
| (b)
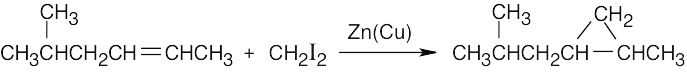
|
Depending on the stereochemistry of the double bond of the alkene in (b), two different isomers can be formed. |
| 8.18 |
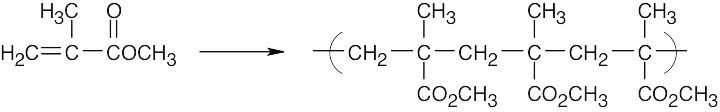
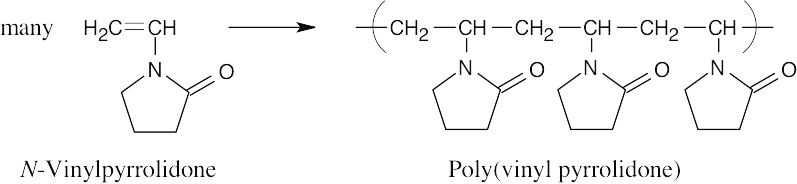
Find the smallest repeating unit in each polymer and add a double bond. This is the monomer unit.
| Monomer Polymer |
| (a)
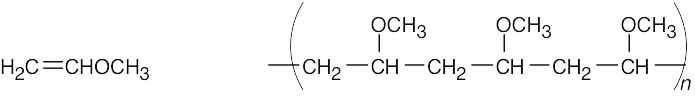
|
| (b)
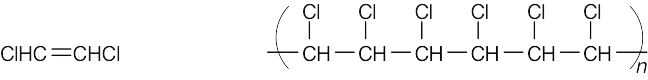
|
|
| 8.19 |
One radical abstracts a hydrogen atom from a second radical, and the remaining two electrons create a double bond.
|
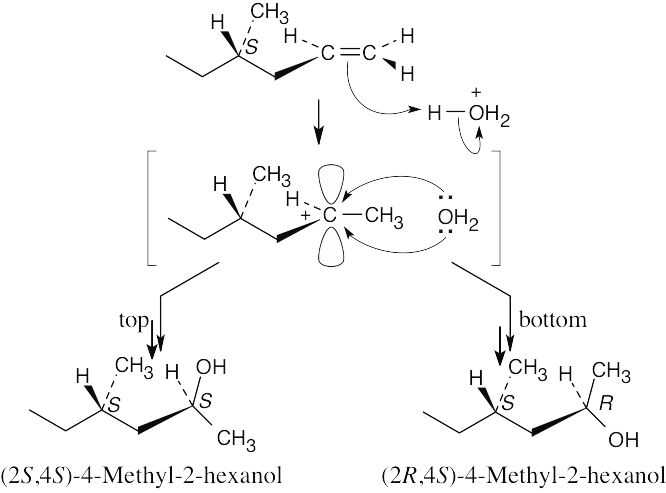
| 8.20 |
Look back to Figure 8.13, which shows the reaction of (R)-4-methyl-1-hexene with H3O+. In a similar way, we can write a reaction mechanism for the reaction of H3O+ with (S)-4-methyl-1-hexene.
The products shown above are diastereomers and are formed in unequal amounts. The (2S,4S) stereoisomer is the enantiomer of the (2R,4R) isomer (shown in Figure 8.13), and the transition states leading to the formation of these two isomers are enantiomeric and of equal energy. Thus, the (2S,4S) and (2R,4R) enantiomers are formed in equal amounts. A similar argument can be used to show that the (2R,4S) and (2S,4R) isomers are formed in equal amounts. The product mixture is optically inactive. |
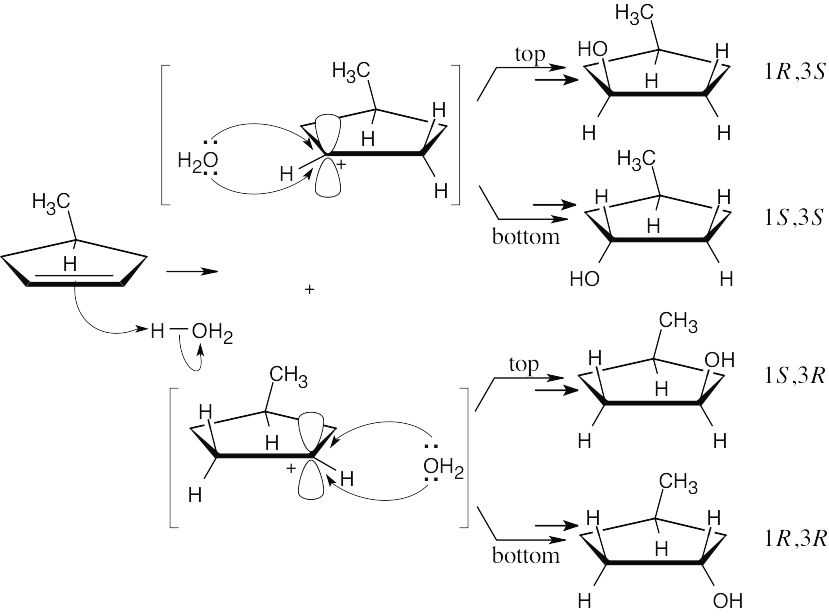
| 8.21 |
Two enantiomeric carbocations are formed. Each carbocation can react with H2O from either the top or the bottom to yield a total of four stereoisomers. The same argument used in Problem 8.20 can be used to show that the (1S,3R) and (1R,3S) enantiomers are formed in equal amounts, and the (1S,3S) and (1R,3R) isomers are formed in equal amounts. The result is a non-50:50 mixture of two racemic pairs. |
Additional Problems
Visualizing Chemistry
| 8.22 |
| (a)
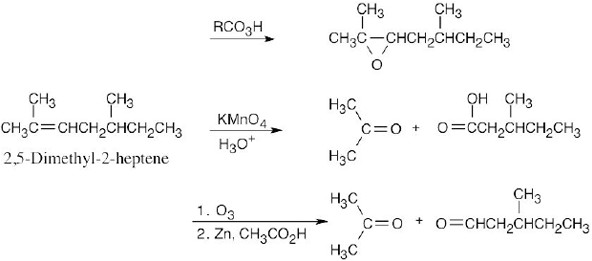
|
| (b)
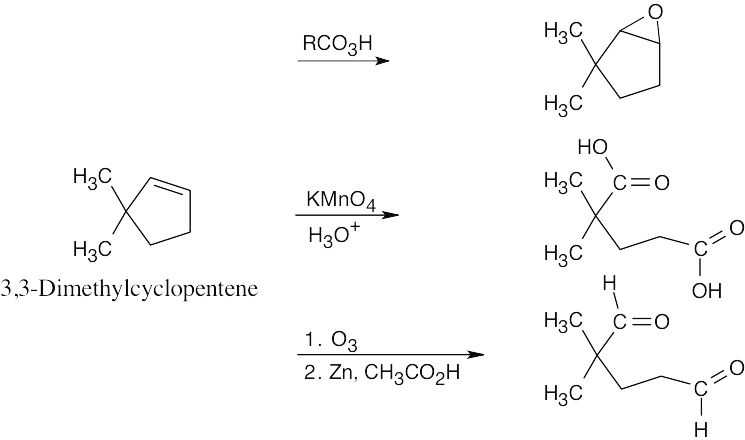
|
|
| 8.23 |
| (a)
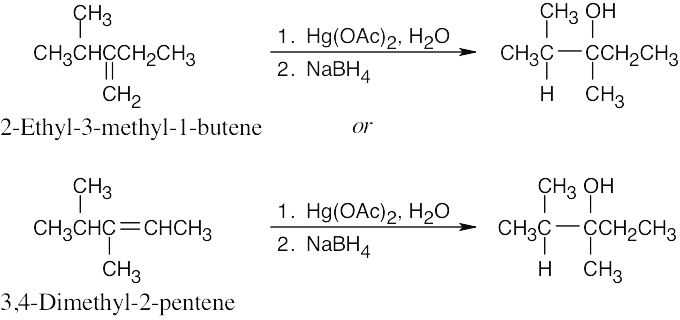
Only oxymercuration/reduction can be used to produce an alcohol that has –OH bonded to the more substituted carbon. A third alkene, 2,3-dimethyl-2-pentene, gives a mixture of tertiary alcohols when treated with either BH3 or Hg(OAc)2. |
| (b)
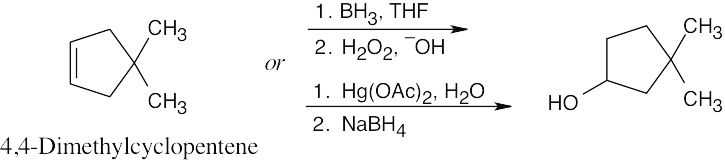
Both hydroboration/oxidation and oxymercuration yield the same alcohol product from the symmetrical alkene starting material. |
|
| 8.24 |
Two possible alcohols might be formed by hydroboration/oxidation of the alkene shown. One product results from addition of BH3 to the top face of the double bond (not formed), and the other product results from addition to the bottom face of the double bond (formed). Addition from the top face does not occur because a methyl group on the bridge of the bicyclic ring system blocks approach of the borane. |
| 8.25 |
RCO3H = meta-Chloroperoxybenzoic acid
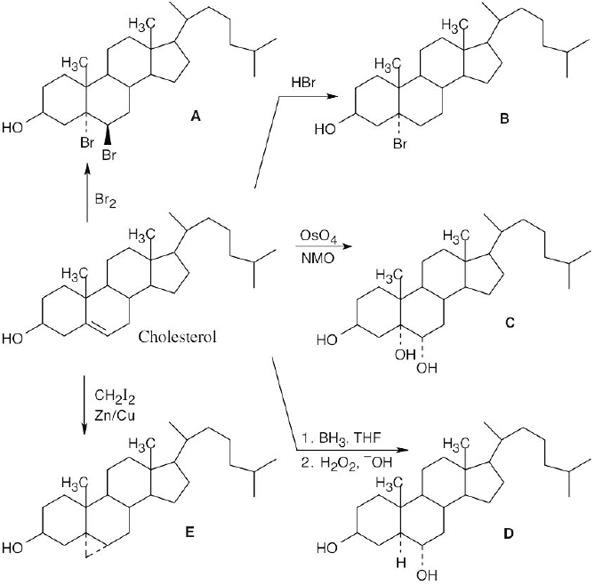
Since the hydroxyl groups in the diol product have a trans relationship, the product can only be formed by epoxide hydrolysis. (Treatment of the alkene with OsO4 yields a product in which the two –OH groups have a cis relationship.) |
Mechanism Problems
| 8.26 |
| (a)

Mechanism:
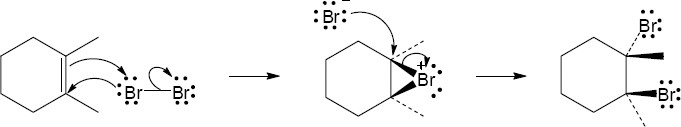
|
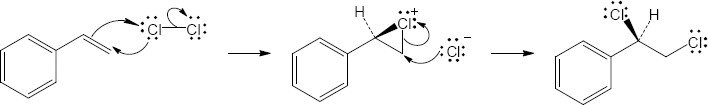
| (b)
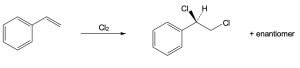
Mechanism:
|
| (c)
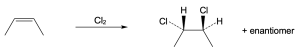
Mechanism:
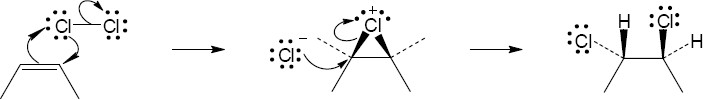
|
|
| 8.27 |
| (a)
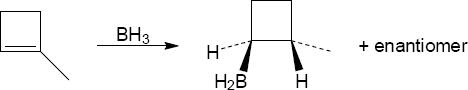
Mechanism:
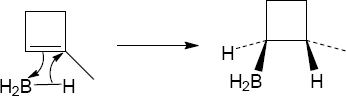
|
| (b)
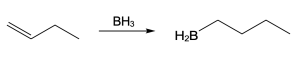
Mechanism:
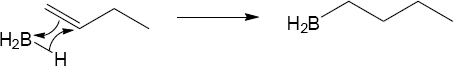
|
| (c)
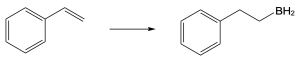
Mechanism:

|
|
| 8.28 |
| (a)
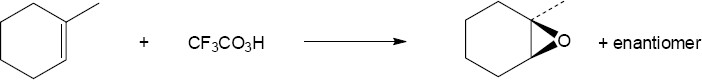
Mechanism:
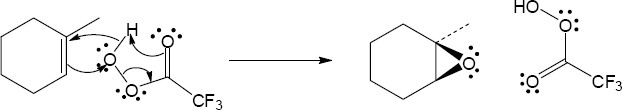
|
| (b)
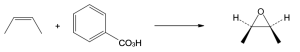
Mechanism:
|
|
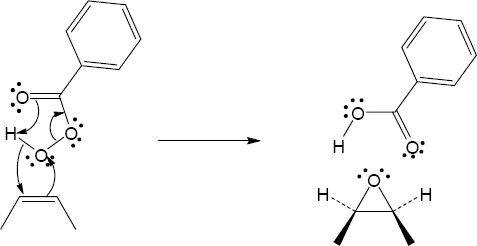
| 8.29 |
| (a)

Mechanism:
|
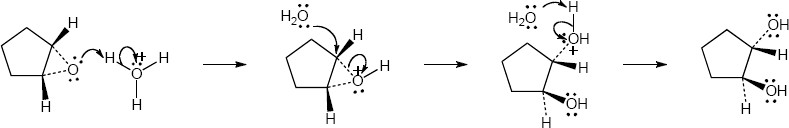
| (b)
Mechanism:
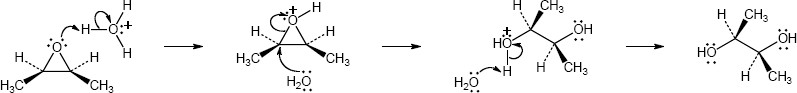
|
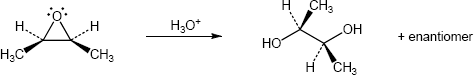
| (c)
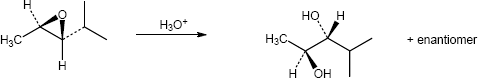
Mechanism:
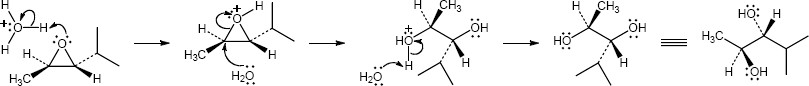
* molecule reoriented with epoxide in the plane of the paper |
|
| 8.30 |
| (a) |
The product of this reaction is achiral (no chiral centers). Therefore, it will not rotate plane polarized light.
|
| (b) |
The product of this reaction is a meso compound. Therefore, it does not rotate plane polarized light.
|
| (c) |
There are two products to this reaction. The first product is a single enantiomer that would rotate plane polarized light, and the second product is achiral (no chiral centers). Therefore, the product mixture would rotate plane polarized light.
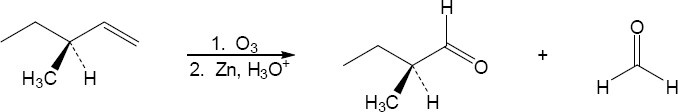
|
|
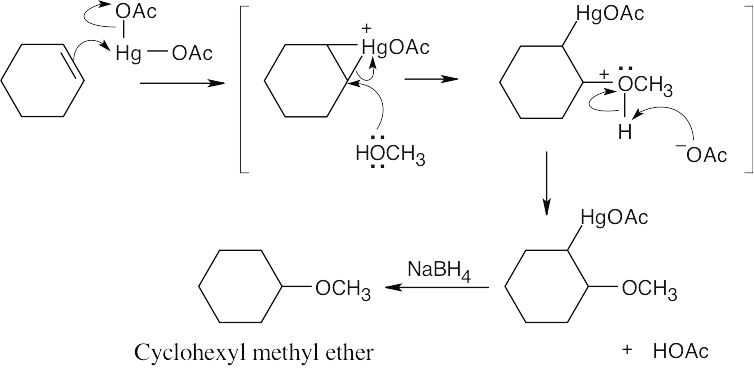
| 8.31 |
Step 1: Protonation of the double bond.
Step 2: Nucleophilic attack of methanol on the carbocation.
Step 3: Loss of proton.
The above mechanism is the same as the mechanism shown in Section 8.4 with one exception: In this problem, methanol, rather than water, is the nucleophile, and an ether, rather than an alcohol, is the observed product. |
| 8.32 |
| (a) |
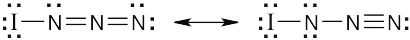 |
| (b) |
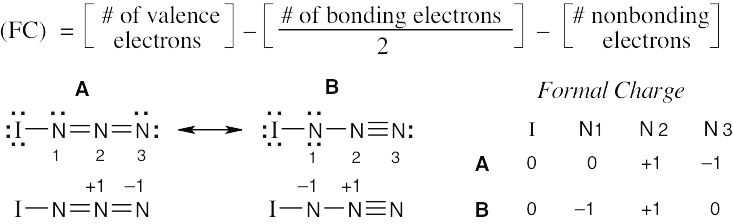
Formal charge calculations show a partial negative charge on N1. |
| (c) |
Addition of IN3 to the alkene yields a product in which –I is bonded to the primary carbon and –N3 is bonded to the secondary carbon. If addition occurs with Markovnikov orientation, I+ must be the electrophile, and the reaction must proceed through an iodonium ion intermediate. Opening of the iodonium ion gives Markovnikov product for the reasons discussed in Problem 8.7. The bond polarity of iodine azide is:
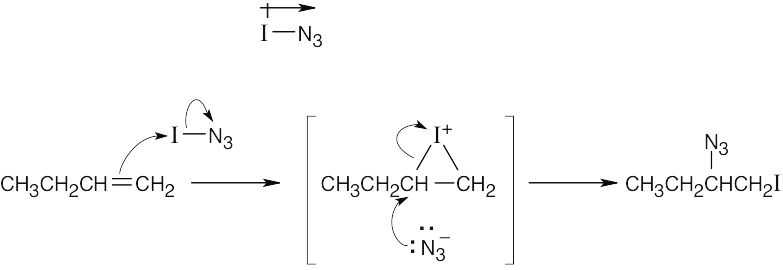
|
|
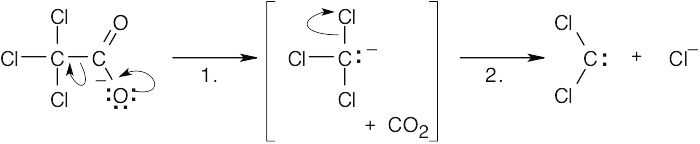
| 8.35 |
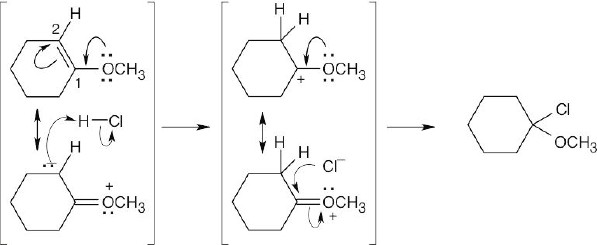
In step 1, carbon dioxide is lost from the trichloroacetate anion. In step 2, elimination of chloride anion produces dichlorocarbene. Step 2 is the same for both the above reaction and the base-induced elimination of HCl from chloroform, and both reactions proceed through the trichloromethanide anion intermediate. |
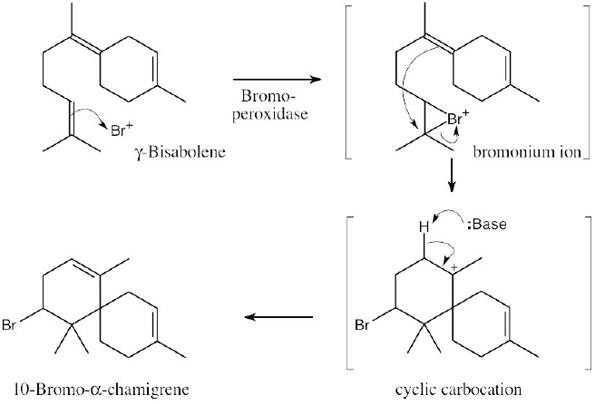
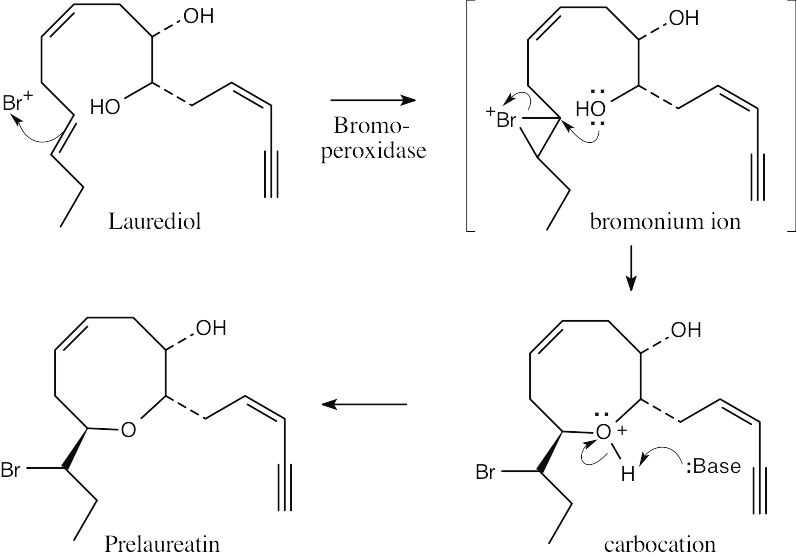
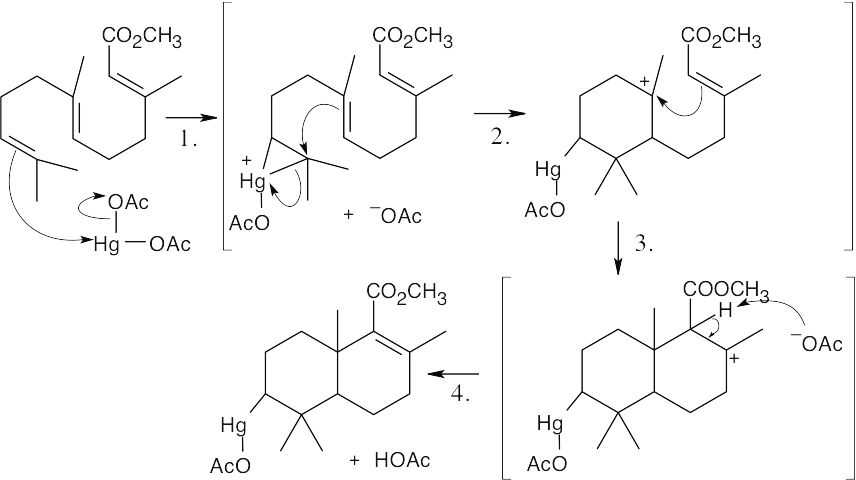
| 8.37 |
The reaction mechanism involves the following steps:
Step 1: Addition of Hg(OAc)2 to one of the double bonds to form a cyclic mercurinium ion.
Step 2: Reaction of a second double bond with the mercurinium ion to form a six-membered ring and a different carbocation.
Step 3: A second cyclization forms the other ring and yields another carbocation.
Step 4: Removal of –H gives a double bond. |
| 8.38 |
Step 1: Formation of a cyclic bromonium ion.
Step 2: Nucleophilic attack of –OH on the bromonium ion.
Step 3: Loss of H+.
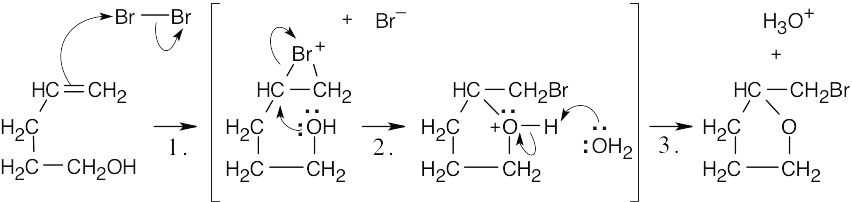
The above mechanism is the same as that for halohydrin formation, shown in Section 8.3. In this case, the nucleophile is the hydroxyl group of 4-penten-1-ol. |
| 8.39 |
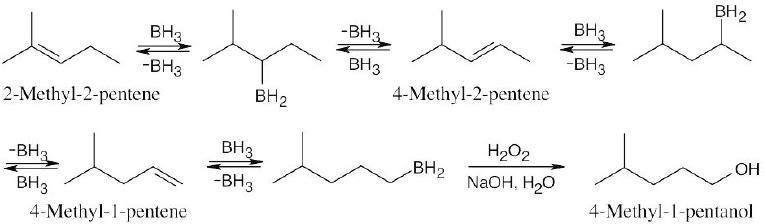
Hydroboration of 2-methyl-2-pentene at 160°C is reversible. The initial organoborane intermediate can eliminate BH3 in either of two ways, yielding either 2-methyl-2-pentene or 4-methyl-2-pentene, which in turn can undergo reversible hydroboration to yield either 4-methyl-2-pentene or 4-methyl-1-pentene. The effect of these reversible reactions is to migrate the double bond along the carbon chain. A final hydroboration then yields the most stable (primary) organoborane, which is oxidized to form 4-methyl-1-pentanol.
|
Reactions of Alkenes
| 8.40 |
| (a)
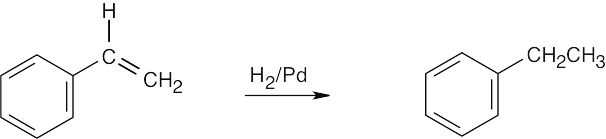
|
| (b)
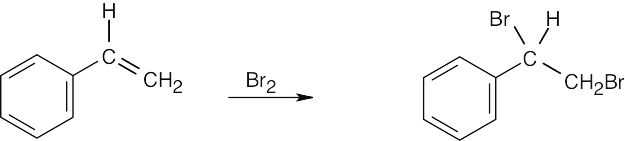
|
| (c)
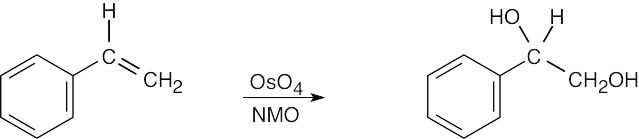
|
| (d)
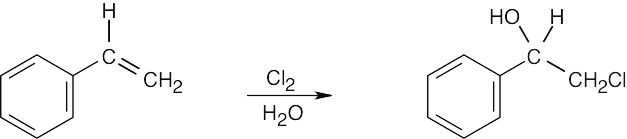
|
| (e)
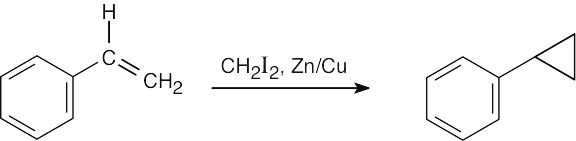
|
| (f)
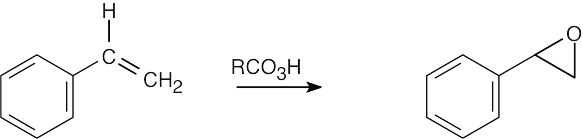
RCO3H = meta-Chloroperoxybenzoic acid |
|
| 8.42 |
| (a)
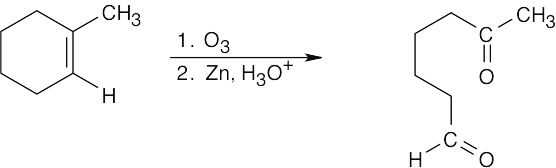
|
| (b)
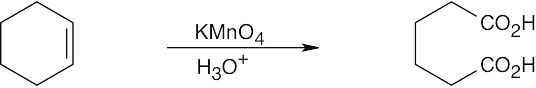
|
| (c)
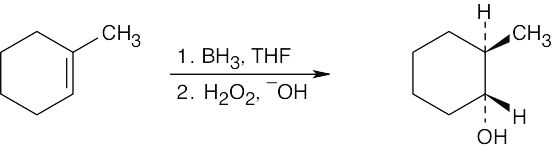
Remember that –H and –OH add syn across the double bond. |
| (d)
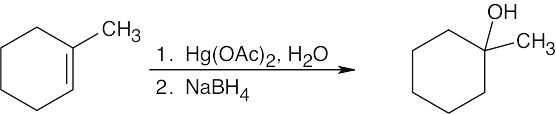
|
|
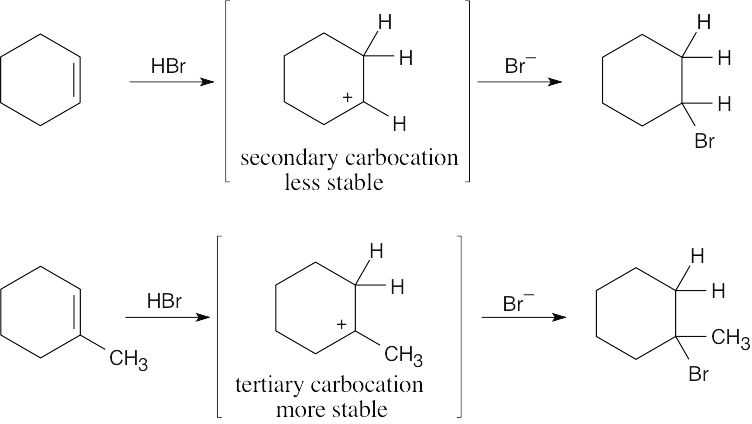
| 8.43 |
Remember from Section 7.10 that a reaction that forms a more stable carbocation intermediate is faster than a comparable reaction that forms a less stable carbocation intermediate. Thus, the reaction of 1-methylcyclohexene with HBr is faster than the reaction of cyclohexene with HBr. |
| 8.44 |
Recall the mechanism of hydroboration and note that the hydrogen added to the double bond comes from borane. The product of hydroboration with BD3 has deuterium bonded to the more substituted carbon; –D and –OH are cis to one another.
|
| 8.45 |
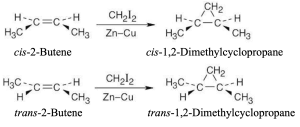
The Simmons–Smith reaction occurs with syn stereochemistry. Only cis-1,2-dimethylcyclopropane is produced from cis-2-butene, and only trans-1,2- dimethylcyclopropane is produced from trans-2-butene. |
| 8.47 |
Conjugation with the Reaction with HCl yields Addition of Cl– leads to
oxygen lone pair electrons a cation intermediate that the observed product.
makes the double bond can be stabilized by the
more nucleophilic. oxygen electrons. |
There are two reasons why the other regioisomer is not formed: (1) Carbon 1 is less nucleophilic than carbon 2; (2) The cation intermediate that would result from protonation at carbon 1 can’t be stabilized by the oxygen electrons. |
Synthesis Using Alkenes
| 8.48 |
| (a)
|
| (b)
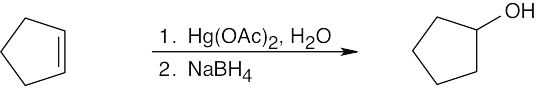
Acid-catalyzed hydration and hydroboration/oxidation are both additional routes to this product. |
| (c)
|
| (d)
|
| (e)
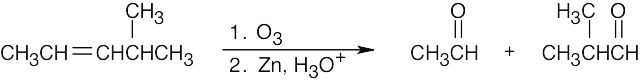
|
| (f)
|
|
| 8.49 |
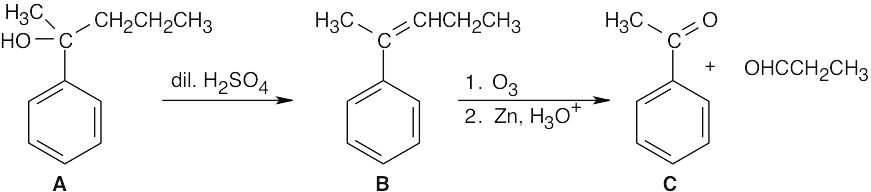
Because ozonolysis gives only one product, we can assume that the alkene is symmetrical.
|
| 8.50 |
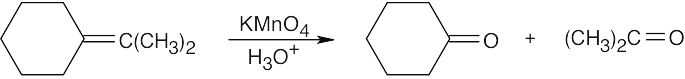
Remember that alkenes can give ketones, carboxylic acids, and CO2 on oxidative cleavage with KMnO4 in acidic solution.
|
| 8.51 |
| (a) Addition of HI occurs with Markovnikov regiochemistry – iodine adds to the more substituted carbon. |
| (b) Hydroxylation of double bonds produces cis, not trans, diols. |
| (c) Ozone reacts with both double bonds of 1,4-cyclohexadiene. |
| (d) Because hydroboration is a syn addition, the –H and the –OH added to the double bond must be cis to each other. |
|
| 8.52 |
| (a) This alcohol can’t be synthesized selectively by hydroboration/oxidation. Consider the two possible starting materials.
1.

1-Pentene yields only the primary alcohol.
2.
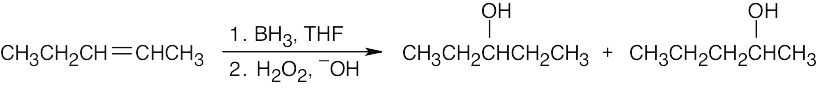
2-Pentene yields a mixture of alcohols. |
| (b)
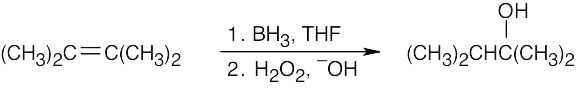
2,3-Dimethyl-2-butene yields the desired alcohol exclusively. |
| (c) This alcohol can’t be formed cleanly by a hydroboration reaction. The –H and –OH added to a double bond must be cis to each other. |
| (d) The product shown is not a hydroboration product; hydroboration yields an alcohol in which –OH is bonded to the less substituted carbon. |
|
Polymers
| 8.55 |
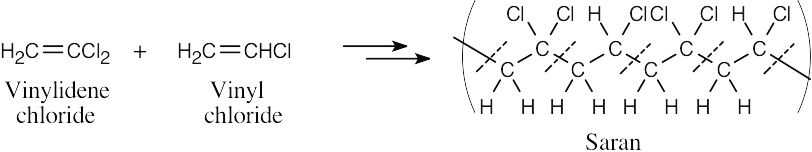
The dashed lines cross the bonds formed during the polymerization reaction. The structural fragments that lie between the dashed lines are the monomer units. Saran is a copolymer of vinylidene chloride and vinyl chloride. |
General Problems
| 8.56 |
| (a) |
Compound A has three degrees of unsaturation. Because compound A contains only one double bond, the other two degrees of unsaturation must be rings. |
| (b), (c) |
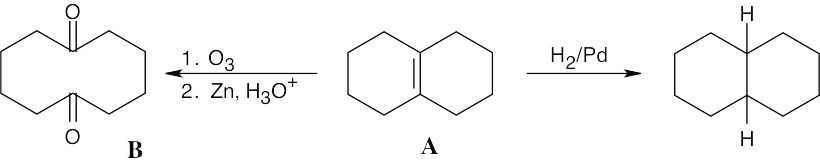 |
Other compounds containing two fused rings and a shared double bond also yield symmetrical diketone products. |
| 8.57 |
(1) |
Hydrocarbon A (C6H12) has one double bond or ring. |
|
(2) |
Because A reacts with one equivalent of H2, it has one double bond and no ring. |
|
(3) |
Compound A forms a diol (B) when reacted with OsO4. |
|
(4) |
When alkenes are oxidized with KMnO4 they give either carboxylic acids or ketones, depending on the substitution pattern of the double bond.
(a) A ketone is produced from what was originally a disubstituted carbon in the double bond.
(b) A carboxylic acid is produced from what was originally a monosubstituted carbon in the double bond. |
|
(5) |
One fragment from KMnO4 oxidation is a carboxylic acid, CH3CH2CO2H.
(a) This fragment was CH3CH2CH= (a monosubstituted double bond) in compound A.
(b) It contains three of the six carbons of compound A. |
|
(6) |
(a) The other fragment contains three carbons.
(b) It forms ketone C on oxidation.
(c) The only three carbon ketone is acetone, O=C(CH3)2.
(d) This fragment was =C(CH3)2 in compound A. |
|
(7) |
If we join the fragment in 5(a) with the one in 6(d), we get:
CH3CH2CH = C(CH3)2 C6H12
A
The complete scheme:
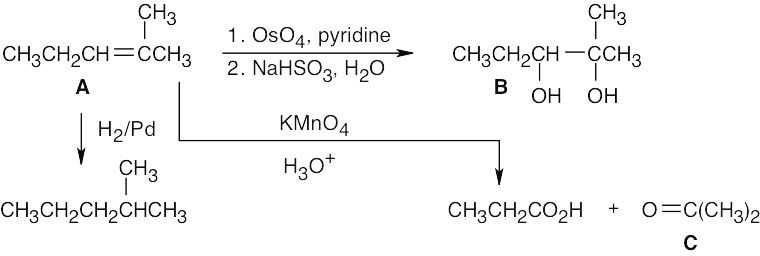
|
| 8.58 |
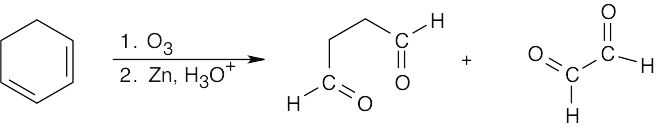
The oxidative cleavage reaction of alkenes with O3, followed by Zn in acid, produces aldehyde and ketone functional groups at sites where double bonds used to be. On ozonolysis, these two dienes yield only aldehydes because all double bonds are monosubstituted.
Because the other diene is symmetrical, only one dialdehyde, OCHCH2CHO, is produced. |
| 8.59 |
Try to solve this problem phrase by phrase.
| (1) C10H18O has two double bonds and/or rings. |
| (2) C10H18O must be an alcohol because it undergoes reaction with H2SO4 to yield an alkene. |
| (3) When C10H18O is treated with dilute H2SO4, a mixture of alkenes of the formula C10H16 is produced. |
| (4) Since the major alkene product B yields only cyclopentanone, C5H8O, on ozonolysis, B and A contain two rings. A therefore has no double bonds.
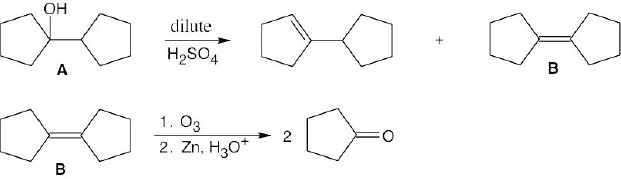
|
|
| 8.61 |
Focus on the stereochemistry of the three-membered ring. Simmons–Smith reaction of 1,1-diiodoethane with the double bond occurs with syn stereochemistry and can produce two isomers. In one of these isomers (A), the methyl group is on the same side of the three- membered ring as the cyclohexane ring carbons. In B, the methyl group is on the side of the three-membered ring opposite to the cyclohexane ring carbons. |
| 8.63 |
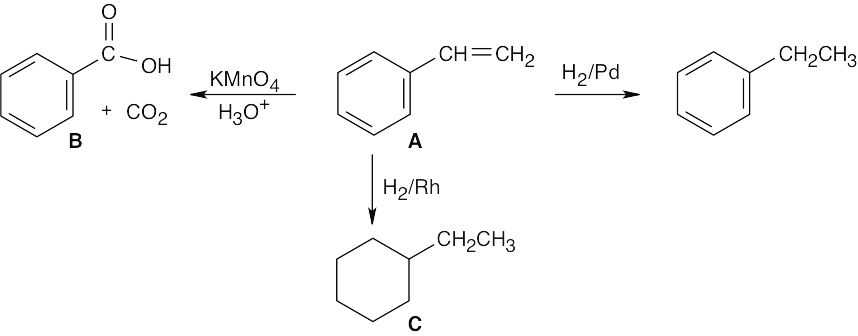
C8H8 has five double bonds and/or rings. One of these double bonds reacts with H2/Pd. Stronger conditions cause the uptake of four equivalents of H2. C8H8 thus contains four double bonds, three of which are in an aromatic ring, and one C=C double bond. A good guess for C8H8 at this point is:
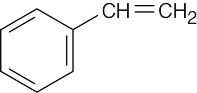
Reaction of a double bond with KMnO4 yields cleavage products of the highest possible degree of oxidation. In this case, the products are CO2 + C6H5CO2H.
|
| 8.64 |
(a) |
Bromine dissolved in CH2Cl2 has a reddish-brown color. When an alkene such as cyclopentene is added to the bromine solution, the double bond reacts with bromine, and the color disappears. This test distinguishes cyclopentene from cyclopentane, which does not react with Br2. Alternatively, each compound can be treated with H2/Pd. The alkene takes up H2, and the alkane is unreactive. |
|
(b) |
An aromatic compound such as benzene is unreactive to the Br2/CH2Cl2 reagent and can be distinguished from 2-hexene, which decolorizes Br2/CH2Cl2. Also, an aromatic compound doesn’t take up H2 under reaction conditions used for hydrogenation of alkenes. |
| 8.65 |
(a) |
α-Terpinene, C10H16, has three degrees of unsaturation. |
|
(b) |
Hydrogenation removes only two degrees of saturation, producing a hydrocarbon C10H20, that has one ring. α-Terpinene thus has two double bonds and one ring. |
|
(c) |
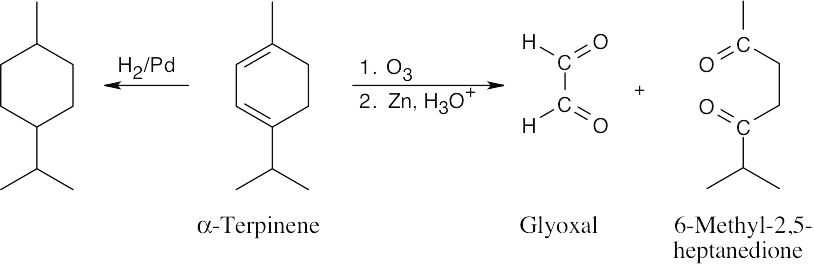 |
| 8.66 |
The models of the cis and trans diols show that it is much easier to form a five-membered cyclic periodate from the cis diol A than from the trans diol B. The cis periodate intermediate is of lower energy than the trans periodate intermediate because of the lack of strain in the cis periodate ring. Because any factor that lowers the energy of a transition state or intermediate also lowers ΔG‡ and increases the rate of reaction, diol cleavage should proceed more slowly for trans diols than for cis diols. |
| 8.67 |
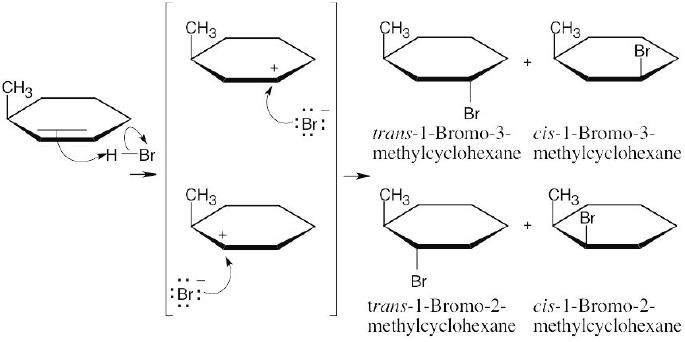
In the reaction of 3-methylcyclohexene with HBr, two intermediate carbocations of approximately equal stability are formed. Both react with bromide ion from top and bottom faces to give four different products.
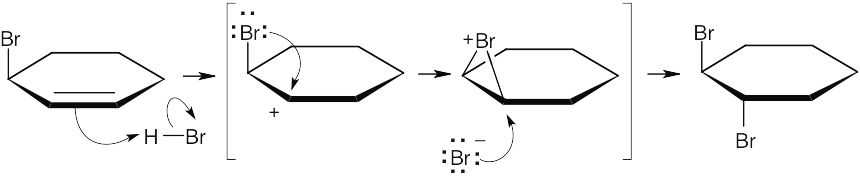
The most stable cation intermediate from protonation of 3-bromocyclohexene is a cyclic bromonium ion, which is attacked by Br– from the opposite side to yield trans product. |
|
| 8.68 |
Addition of one equivalent of HX or X2 to a triple bond occurs with Markovnikov regiochemistry to yield a product in which the two added atoms usually have a trans relationship across the double bond. |
| 8.69 |
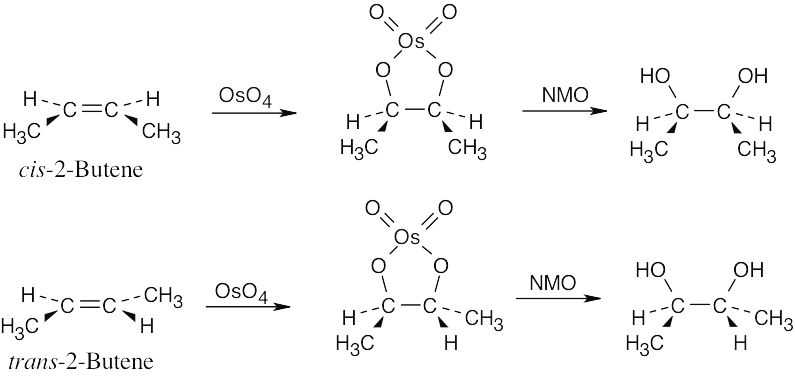
Formation of the cyclic osmate, which occurs with syn stereochemistry, retains the cis-trans stereochemistry of the double bond because osmate formation is a single-step reaction.
Oxidation of the osmate does not affect the stereochemistry of the carbon–oxygen bond, and the diol produced from cis-2-butene is a stereoisomer of the diol produced from trans-2-butene. |
| 8.70 |
A has four multiple bonds/rings.
2-Phenyl-3-pentanol is also an acceptable answer. |
This file is copyright 2023, Rice University. All Rights Reserved.