
25.6 Reactions of Monosaccharides
Because monosaccharides have only two kinds of functional groups, hydroxyls and carbonyls, most of the chemistry of monosaccharides is the familiar chemistry of these two groups. As we’ve seen, alcohols can be converted into esters and ethers and can be oxidized; carbonyl compounds can react with nucleophiles and can be reduced.
Ester and Ether Formation
Monosaccharides behave as simple alcohols in much of their chemistry. For example, carbohydrate –OH groups can be converted into esters and ethers, which are often easier to work with than the free sugars. Because of their many hydroxyl groups, monosaccharides are usually soluble in water but insoluble in organic solvents such as ether. They are also difficult to purify and have a tendency to form syrups rather than crystals when water is removed. Ester and ether derivatives, however, are soluble in organic solvents and are easily purified and crystallized.
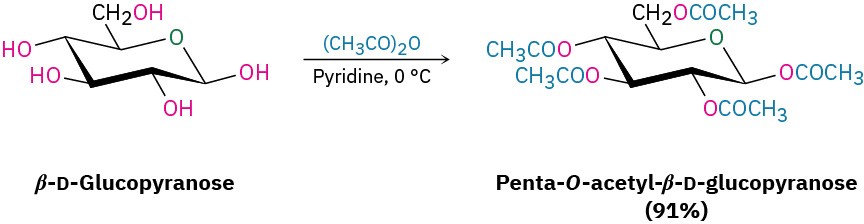
Esterification is normally carried out by treating a carbohydrate with an acid chloride or acid anhydride in the presence of a base (Section 21.4 and Section 21.5). All the –OH groups react, including the anomeric one. For example, β-D-glucopyranose is converted into its pentaacetate by treatment with acetic anhydride in pyridine solution.
Carbohydrates are converted into ethers by treatment with an alkyl halide in the presence of base—the Williamson ether synthesis (Section 18.2). Standard Williamson conditions using a strong base tend to degrade sensitive sugar molecules, but silver oxide works well as a mild base and gives high yields of ethers. For example, α-D-glucopyranose is converted into its pentamethyl ether in 85% yield on reaction with iodomethane and Ag2O.
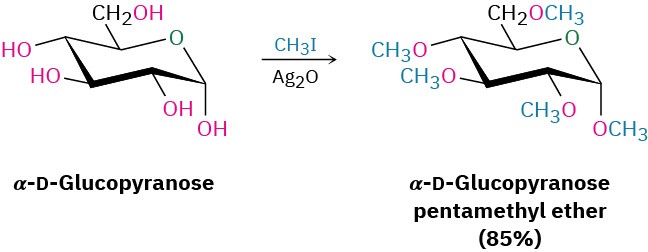
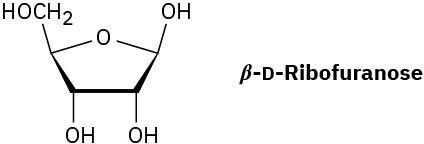
Problem 25-16
Draw the products you would obtain by reaction of β-D-ribofuranose with: (a) CH3I, Ag2O
(b) (CH3CO)2O, pyridine
Glycoside Formation
We saw in Section 19.10 that treatment of a hemiacetal with an alcohol and an acid catalyst yields an acetal.
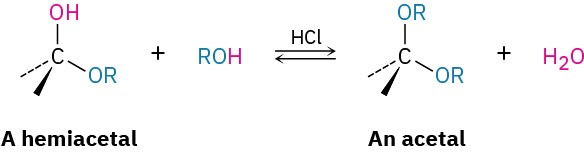
In the same way, treatment of a monosaccharide hemiacetal with an alcohol and an acid catalyst yields an acetal called a glycoside, in which the anomeric –OH has been replaced by an –OR group. For example, reaction of β-D-glucopyranose with methanol gives a mixture of α and β methyl D-glucopyranosides. (Note that a glycoside is the functional group name for any sugar, whereas a glucoside is formed specifically from glucose.)
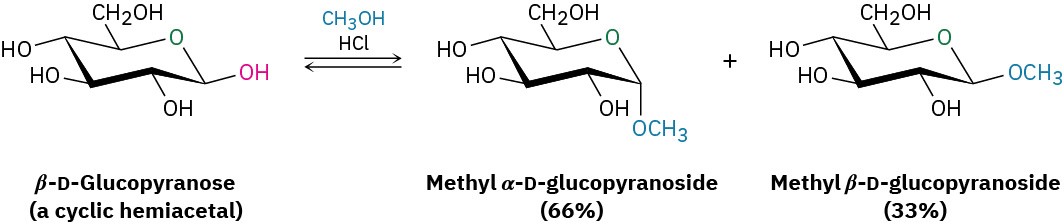
Glycosides are named by first citing the alkyl group and then replacing the –ose ending of the sugar with –oside. Like all acetals, glycosides are stable in neutral water. They aren’t in equilibrium with an open-chain form, and they don’t show mutarotation. They can, however, be hydrolyzed to give back the free monosaccharide plus alcohol on treatment with aqueous acid (Section 19.10).
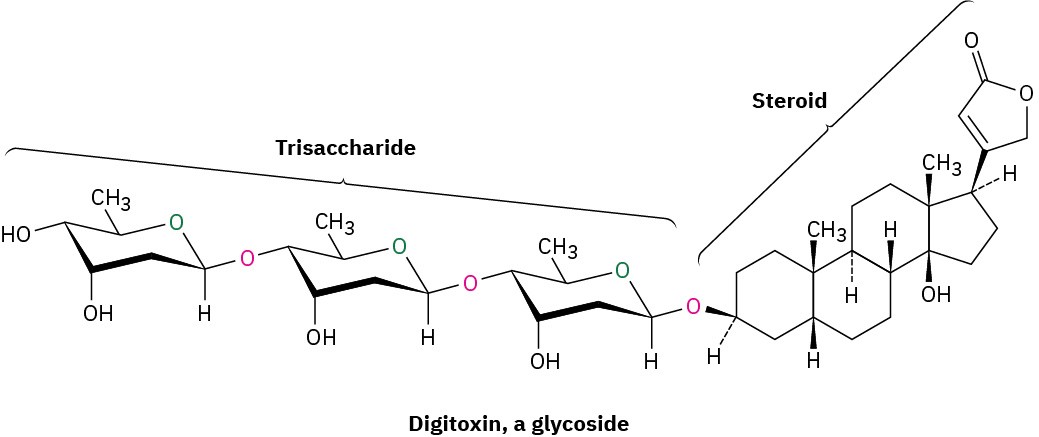
Glycosides are abundant in nature, and many biologically important molecules contain glycosidic linkages. For example, digitoxin, the active component of the digitalis preparations used for treatment of heart disease, is a glycoside consisting of a steroid alcohol linked to a trisaccharide. Note also that the three sugars are linked to one another by glycoside bonds.
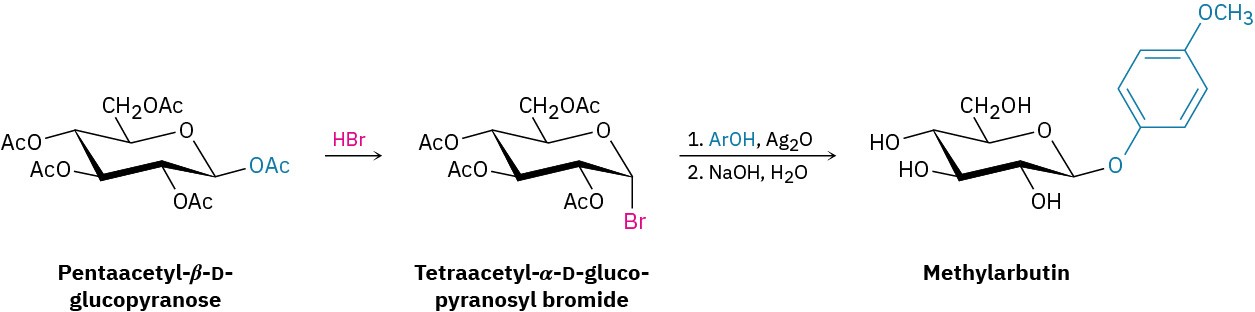
The laboratory synthesis of glycosides can be difficult because of the numerous –OH groups on the sugar molecule. One method that is particularly suitable for preparing glucose β– glycosides involves treatment of glucose pentaacetate with HBr, followed by addition of the appropriate alcohol in the presence of silver oxide. Called the Koenigs–Knorr reaction, the sequence involves formation of a pyranosyl bromide, followed by nucleophilic substitution. For example, methylarbutin, a glycoside found in pears, has been prepared by reaction of tetraacetyl-α-D-glucopyranosyl bromide with p-methoxyphenol.
Although the Koenigs–Knorr reaction appears to involve a simple backside SN2 displacement of bromide ion by alkoxide ion, the situation is actually more complex. Both α and β anomers of tetraacetyl-D-glucopyranosyl bromide give the same β-glycoside product, implying that they react by a common pathway.
This result can be understood by assuming that tetraacetyl-D-glucopyranosyl bromide (either α or β anomer) undergoes a spontaneous SN1-like loss of Br–, followed by internal reaction with the ester group at C2 to form an oxonium ion. Since the acetate at C2 is on the bottom of the glucose ring, the C–O bond also forms from the bottom. Backside SN2 displacement of the oxonium ion then occurs with the usual inversion of configuration, yielding a β-glycoside and regenerating the acetate at C2 (Figure 25.7).
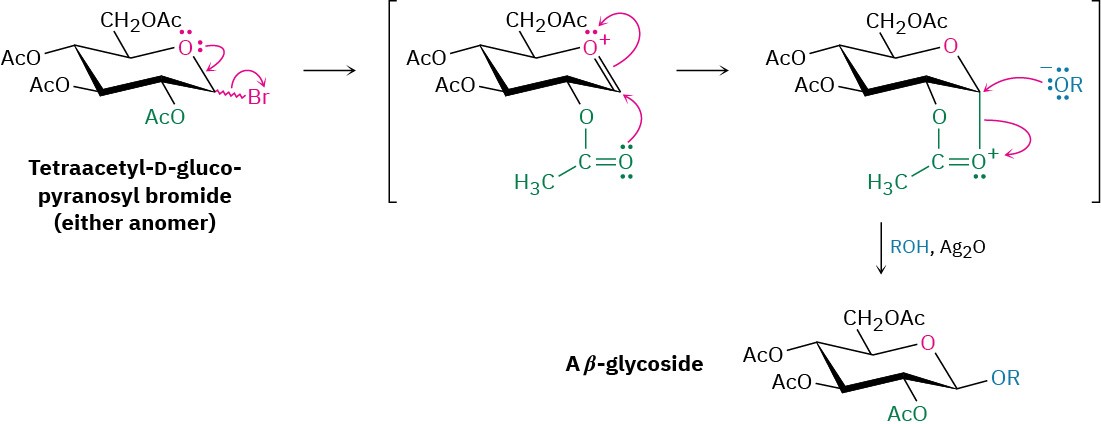
Figure 25.7Mechanism of the Koenigs–Knorr reaction, showing the neighboring- group effect of a nearby acetate.
The participation shown by the nearby acetate group in the Koenigs–Knorr reaction is referred to as a neighboring-group effect and is a common occurrence in organic chemistry. Neighboring-group effects are usually noticeable only because they affect the rate or stereochemistry of a reaction; the nearby group itself does not undergo any evident change during the reaction.
Biological Ester Formation: Phosphorylation
In living organisms, carbohydrates occur not only in the free form but also linked through their anomeric center to other molecules such as lipids (glycolipids) or proteins (glycoproteins). Collectively called glycoconjugates, these sugar-linked molecules are components of cell walls that are crucial to the mechanism by which different cell types recognize one another.
Glycoconjugate formation occurs by reaction of the lipid or protein with a glycosyl nucleoside diphosphate. This diphosphate is itself formed by initial reaction of a monosaccharide with adenosine triphosphate (ATP) to give a glycosyl monophosphate, followed by reaction with uridine triphosphate (UTP), to give a glycosyl uridine diphosphate. (We’ll see the structures of nucleoside phosphates in Section 28.1.) The purpose of the phosphorylation is to activate the anomeric –OH group of the sugar and make it a better leaving group in a nucleophilic substitution reaction by a protein or lipid (Figure 25.8).
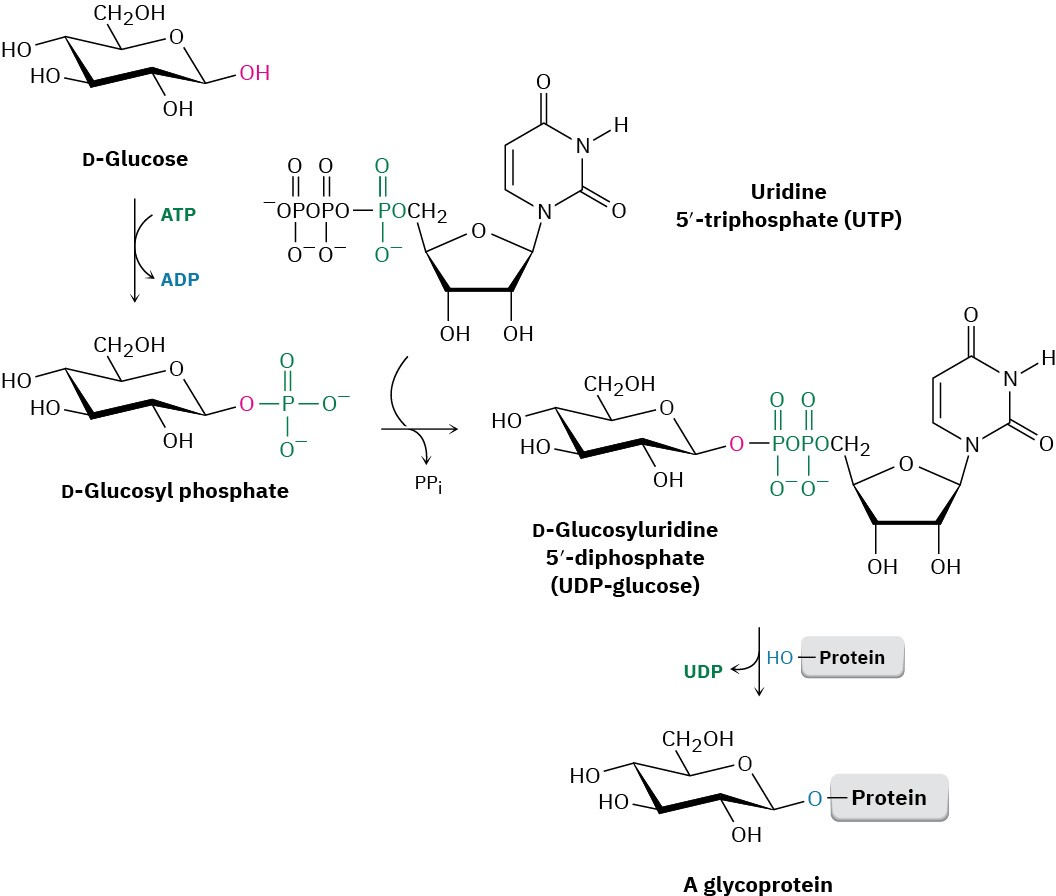
Figure 25.8 Glycoprotein formation occurs by initial phosphorylation of the starting carbohydrate with ATP to a glycosyl monophosphate, followed by reaction with UTP to form a glycosyl uridine 5′-diphosphate. Nucleophilic substitution by an –OH (or – NH2) group on a protein then gives the glycoprotein.
Reduction of Monosaccharides
Treatment of an aldose or ketose with NaBH4 reduces it to a polyalcohol called an alditol. The reduction occurs by reaction of the open-chain form present in the aldehyde/ketone ⇄ hemiacetal equilibrium. Although only a small amount of the open-chain form is present at any given time, that small amount is reduced, more is produced by opening of the pyranose form, that additional amount is reduced, and so on, until the entire sample has undergone reaction.
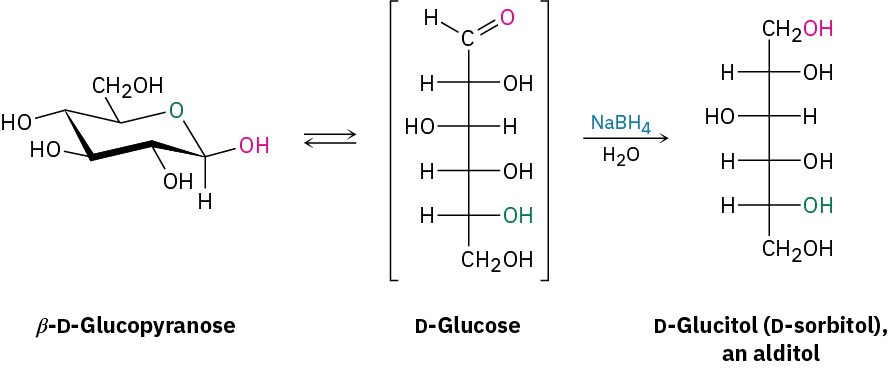
D-Glucitol, the alditol produced by reduction of D-glucose, is itself a naturally occurring substance found in many fruits and berries. It is used under the name D-sorbitol as a sweetener and sugar substitute in many foods.
Problem 25-17
Reduction of D-glucose leads to an optically active alditol (D-glucitol), whereas reduction of D-galactose leads to an optically inactive alditol. Explain.
Problem 25-18
Reduction of L-gulose with NaBH4 leads to the same alditol (D-glucitol) as reduction of D- glucose. Explain.
Oxidation of Monosaccharides
Like other aldehydes, aldoses are easily oxidized to yield the corresponding carboxylic acids, called aldonic acids. A buffered solution of aqueous Br2 is often used for this purpose.
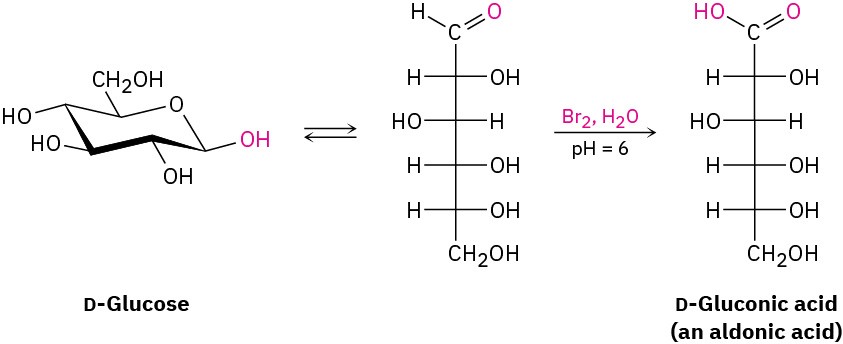
Historically, the oxidation of an aldose with either Ag+ in aqueous ammonia (called Tollens’ reagent) or Cu2+ with aqueous sodium citrate (Benedict’s reagent) formed the basis of simple tests for what are called reducing sugars. (Reducing because the aldose reduces the metal oxidizing agent.) Some simple diabetes self-test kits sold in drugstores still use Benedict’s reagent to detect glucose in urine, though more modern methods have largely replaced it.
All aldoses are reducing sugars because they contain an aldehyde group, but some ketoses are reducing sugars as well. Fructose reduces Tollens’ reagent, for example, even though it
contains no aldehyde group. Reduction occurs because fructose is readily isomerized to a mixture of aldoses (glucose and mannose) in basic solution by a series of keto–enol tautomeric shifts (Figure 25.9). Glycosides, however, are nonreducing because the acetal group is not hydrolyzed to an aldehyde under basic conditions.
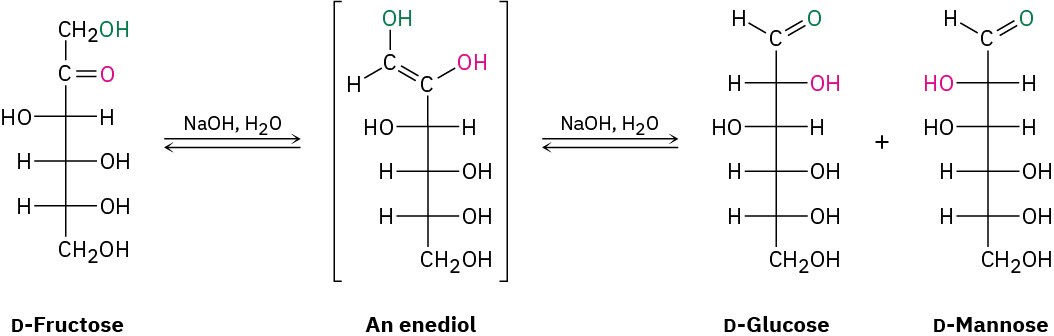
Figure 25.9Fructose, a ketose, is a reducing sugar because it undergoes two base- catalyzed keto–enol tautomerizations that result in conversion to a mixture of aldoses.
If warm, dilute HNO3 (nitric acid) is used as the oxidizing agent, an aldose is oxidized to a dicarboxylic acid called an aldaric acid. Both the aldehyde carbonyl and the terminal – CH2OH group are oxidized in this reaction.
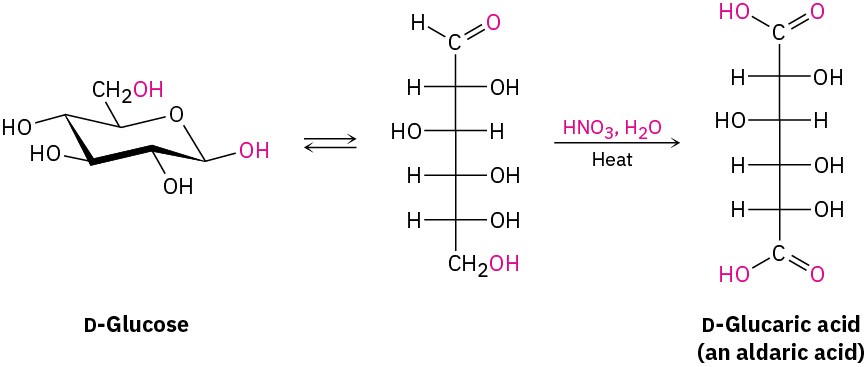
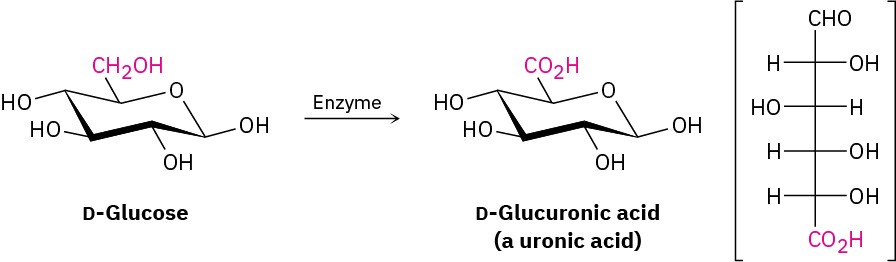
Finally, if only the –CH2OH end of the aldose is oxidized without affecting the –CHO group, the product is a monocarboxylic acid called a uronic acid. The reaction can only be done enzymatically; no chemical reagent is known that can accomplish this selective oxidation in the laboratory.
Problem 25-19
D-Glucose yields an optically active aldaric acid on treatment with HNO3, but D-allose yields an optically inactive aldaric acid. Explain.
Problem 25-20
Which of the other six D aldohexoses yield optically active aldaric acids on oxidation, and which yield optically inactive (meso) aldaric acids? (See Problem 25-19.)
Chain Lengthening: The Kiliani–Fischer Synthesis
Much early activity in carbohydrate chemistry was devoted to unraveling the stereochemical relationships among monosaccharides. One of the most important methods used was the Kiliani–Fischer synthesis, which results in the lengthening of an aldose chain by one carbon atom. The C1 aldehyde group of the starting sugar becomes C2 of the chain- lengthened sugar, and a new C1 carbon is added. For example, an aldopentose is converted by Kiliani–Fischer synthesis into two aldohexoses.
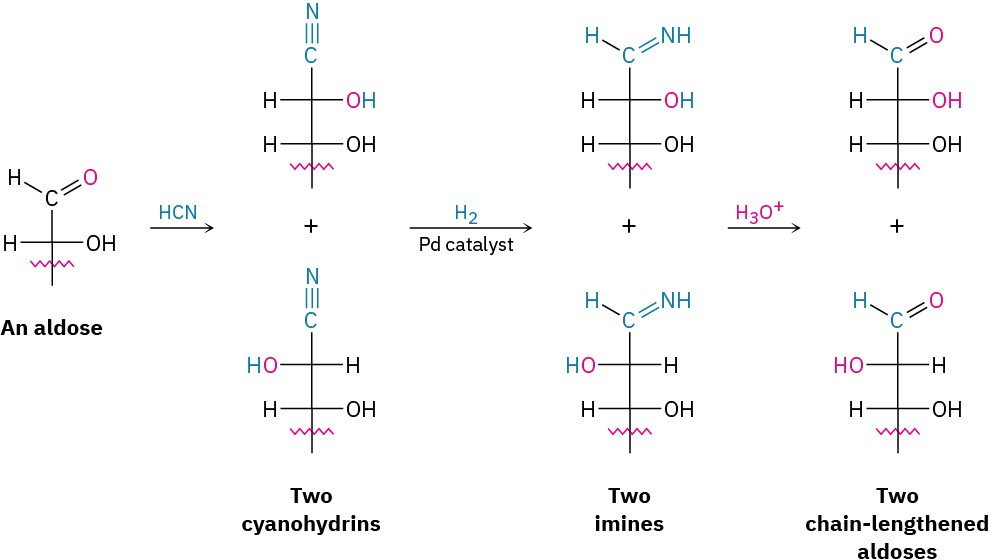
Discovery of the chain-lengthening sequence was initiated by the observation of Heinrich Kiliani in 1886 that aldoses react with HCN to form cyanohydrins (Section 19.6). Emil Fischer immediately realized the importance of Kiliani’s discovery and devised a method for converting the cyanohydrin nitrile group into an aldehyde.
Fischer’s original method for conversion of the nitrile into an aldehyde involved hydrolysis to a carboxylic acid, ring closure to a cyclic ester (lactone), and subsequent reduction. A modern improvement involves reducing the nitrile over a palladium catalyst, yielding an imine intermediate that is hydrolyzed to an aldehyde. Note that the cyanohydrin is formed as a mixture of stereoisomers at the new chirality center, so two new aldoses, differing only in their stereochemistry at C2, result from Kiliani–Fischer synthesis. Chain extension of D- arabinose, for example, yields a mixture of D-glucose and D-mannose.
Problem 25-21
What product(s) would you expect from Kiliani–Fischer reaction of D-ribose?
Problem 25-22
What aldopentose would give a mixture of L-gulose and L-idose on Kiliani–Fischer chain extension?
Chain Shortening: The Wohl Degradation
Just as the Kiliani–Fischer synthesis lengthens an aldose chain by one carbon, the Wohl degradation shortens an aldose chain by one carbon. Wohl degradation is almost the exact opposite of the Kiliani–Fischer sequence. That is, the aldose aldehyde carbonyl group is first converted into a nitrile, and the resulting cyanohydrin loses HCN under basic conditions—the reverse of a nucleophilic addition reaction.
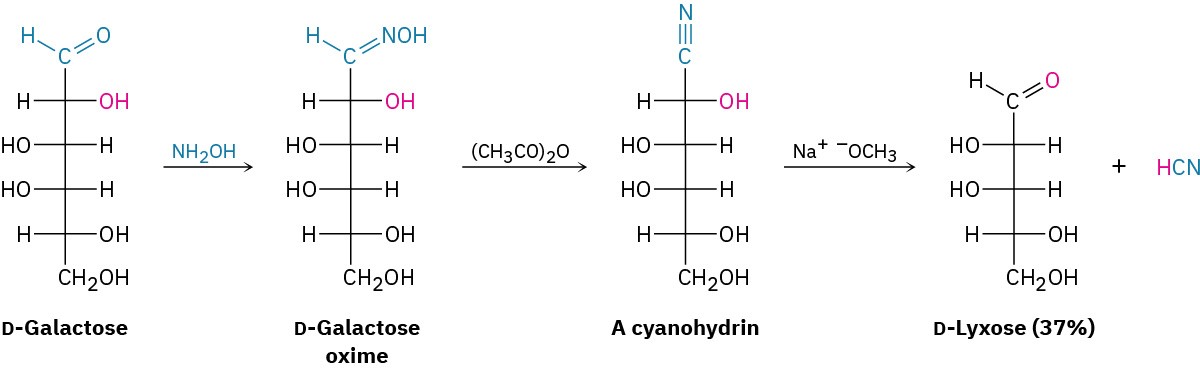
Conversion of the aldehyde into a nitrile is accomplished by treatment of an aldose with hydroxylamine to give an imine called an oxime (Section 19.8), followed by dehydration of the oxime with acetic anhydride. The Wohl degradation does not give particularly high yields of chain-shortened aldoses, but the reaction is general for all aldopentoses and aldohexoses. For example, D-galactose is converted by Wohl degradation into D-lyxose.
Problem 25-23
Two of the four D aldopentoses yield D-threose on Wohl degradation. What are their structures?
